- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Assessment of RNA Polymerase I Activity

Published: Vol 7, Iss 3, Feb 5, 2017 DOI: 10.21769/BioProtoc.2120 Views: 9684
Reviewed by: HongLok LungGaston A. PizzioAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
In eukaryotic cells transcriptional processes are carried out by three different RNA polymerases: RNA polymerase I which specifically transcribes ribosomal RNA (rRNA), RNA polymerase II which transcribes protein-coding genes to yield messenger RNAs (mRNAs) and small RNAs, while RNA polymerase III transcribes the genes for transfer RNAs and for the smallest species of ribosomal RNA (5S rRNA). This protocol describes an in vitro assay to evaluate the rRNA transcriptional activity of RNA polymerase I. The method measures the quantity of radiolabelled uridine 5’ triphosphate incorporated in ex novo synthesized rRNA molecules by RNA polymerase I, in optimal conditions for the enzyme activity and in the presence of a toxin, α-amanitin, which inhibits RNA polymerase II and III without affecting RNA polymerase I (Novello and Stirpe, 1970).
Keywords: RNA polymerase IBackground
In eukaryotic cells the RNA polymerase I transcribes ribosomal genes, which are located in the nucleolus, producing 45S rRNA precursor molecules. These are processed to form the mature 18S, 5.8S and 28S rRNA. They are essential for the assembly of the 60S and the 40S subunits of mature ribosomes. Recent evidence indicates that the ribosome biogenesis rate is related to cell cycle length (Derenzini et al., 2005) and may play a role in tumorigenesis by controlling the expression of the tumour suppressor protein p53. Cells with an up-regulated ribosome biogenesis are rapidly proliferating and are characterized by a down-regulated p53 expression (Donati et al., 2011). Moreover, the ribosome biogenesis rate influences the sensitivity of cancer cells to chemotherapeutic agents which hinder rRNA transcription: higher the rate of ribosome biogenesis, higher the cytotoxic effect induced by the drug (Scala et al., 2016). Therefore, the evaluation of the ribosome biogenesis rate will become a more and more utilized procedure both in tumour pathology and in clinical oncology (Montanaro et al., 2013). Since the rate of ribosome production is tightly conditioned by the rate of 45S precursor molecules transcription, all the methods used for the evaluation of ribosome biogenesis rate measure the synthesized 45S rRNA. The used methods are: quantitative evaluation of 45S rRNA transcripts by real time PCR analysis; quantitative analysis of 45S rRNA, separated by gel electrophoresis of total RNA extracted from cells labelled with 32P-orthophosphate, and visualized by autoradiography; and quantitative evaluation of radiolabelled uridine 5’ triphosphate incorporated in ex novo synthesized rRNA molecules by RNA polymerase I. The first two methods measure the quantity of 45S rRNA present in the cells, that may be influenced by changes of rRNA processing mechanism, whereas the method described here quantifies the transcriptional activity of the RNA polymerase I and it is indicative of the rRNA transcription rate. This method is very complex and time-consuming and requires special accuracy, but it is still the only one method to selectively measure the rRNA transcription rate.
Materials and Reagents
- Cell scraper, 40 cm handle, 16 mm blade (Sigma-Aldrich, catalog number: C6106 )
- Plastic tubes 50 ml conical base (114 x 28 mm) (SARSTEDT, catalog number: 62.547.254 )
- Plastic tubes 15 ml conical base (120 x 17 mm) (SARSTEDT, catalog number: 62.554.002 )
- Plastic tubes 1.5 ml safe lock (Eppendorf, catalog number: 022363204 )
- Glass microfiber filters, diameter 25 mm, Whatman grade GF/C (GE Healthcare, catalog number: 1822-025 )
- Scintillation plastic vials (Sigma-Aldrich, catalog number: V6755 )
- Microscope slides (26 x 76 mm) (Sigma-Aldrich, catalog number: Z692247 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: S3014 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: P9541 )
- Sodium phosphate dibasic (Na2HPO4) (Sigma-Aldrich, catalog number: S3264 )
- Potassium phosphate monobasic (KH2PO4) (Sigma-Aldrich, catalog number: P9791 )
- Hydrochloric acid (HCl) (Sigma-Aldrich, catalog number: H1758 )
- Tris base (Sigma-Aldrich, catalog number: T1503 )
- Ammonium sulfate [(NH4)2SO4] (Sigma-Aldrich, catalog number: A4418 )
- Sucrose (Merck Millipore, catalog number: 107687 )
- 1 M magnesium chloride (MgCl2) solution (Sigma-Aldrich, catalog number: M1028 )
- Triton X-100 (Sigma-Aldrich, catalog number: T8787 )
- 1 M manganese(II) chloride (MnCl2) solution (Sigma-Aldrich, catalog number: M1787 )
- Guanosine 5’-triphosphate sodium salt (GTP) (Sigma-Aldrich, catalog number: 10106399001 )
- Adenosine 5’-triphosphate disodium salt hydrate (ATP) (Sigma-Aldrich, catalog number: A7699 )
- Cytidine 5’-triphosphate disodium salt (CTP) (Sigma-Aldrich, catalog number: C1506 )
- Uridine 5’-triphosphate tri salt (UTP) (Sigma-Aldrich, catalog number: U6875 )
- Uridine 5’ triphosphate [5,6-3H] tetrasodium salt (3H-UTP) (PerkinElmer, catalog number: NET380250UC )
- α-amanitin (Sigma-Aldrich, catalog number: A2263 )
- Liquid scintillation cocktail Hionic Fluor (PerkinElmer, catalog number: 6013319 )
- Trichloroacetic acid (TCA) (Sigma-Aldrich, catalog number: T4885 )
- Perchloric acid (PCA) 65% w/w (10.35 N) (CARLO ERBA Reagents, catalog number: 306091 )
- Potassium hydroxide (KOH) (EMD Millipore, catalog number: 105033 )
- 2-mercaptoethanol (EMD Millipore, catalog number: 805740 )
- Sodium fluoride (NaF) (Sigma-Aldrich, catalog number: S7920 )
- PBS (pH 7.3) (see Recipe 1)
- 1 M Tris-HCl solution (pH 7.4 or pH 8.0) (see Recipe 2)
- 3 M ammonium sulfate (see Recipe 3)
- Homogenization-solution (see Recipe 4)
- Washing solution (see Recipe 5)
- Suspension solution (see Recipe 6)
- High ionic strength 5x reaction mixture (see Recipe 7)
- Nucleoside triphosphate mixture (see Recipe 8)
- Non-radioactive solution
- Radioactive solution
- α-amanitin reconstitution (see Recipe 9)
- 10% (w/w) TCA (see Recipe 10)
- 0.6 N PCA (see Recipe 11)
- 0.3 N KOH (see Recipe 12)
- 6 N PCA (see Recipe 13)
- 7% (w/w) PCA (see Recipe 14)
- 0.5 M NaF (see Recipe 15)
- Low ionic strength 5x reaction mixture (see Recipe 16)
Equipment
- Protective gloves/protective clothing/eye protection/face protection
- Centrifuge (Beckman Coulter, model: TJ-25 )
Note: This product has been discontinued. - Teflon® pestle PYREX® Potter-Elvehjem tissue grinder (Thomas Scientific, catalog number: 1234F )
- Forceps
- High speed tissue grinder (homogenizer) (IKA, model: Eurostar )
- Inverted microscope (Zeiss, model: Primovert, catalog number: 491206-0001-000 )
- 10 ml glass tubes
- Shaking water bath (JULABO, model: SW22 )
- 2,000 ml filter flask
- Spectrophotometer UV-VIS (CHROMSERVIS, model: Nanogenius )
- Liquid scintillation counter (Guardian, Wallac, PerkinElmer)
- Vacuum glass filter holder for 25 mm disc filters elements: glass funnel, clamp, base for 25 mm glass/filter holder and a perforated stopper (available from many companies, for example: Sigma-Aldrich, catalog number: Z290467 ; EMD Millipore, catalog number: XX1002500 )
- Vacuum pump
Procedure
Note: The experiment utilizes radioactive material and operators require experience in personal and environmental radiation protection and in the management of radioactive waste.
- Cell nuclei isolation
RNA polymerase I activity can be measured in nuclei isolated from cultured cell lines or from fresh or frozen tissues, in this context, the standard procedure of nuclei isolation from cultured cells is described.- Grow the desired cell line in the required culture medium on cell culture flasks until reaching about 50 x 106 cells at least.
Note: Cells may be subjected to specific treatment, grown under special conditions or various cell lines compared. - Harvest cells by scraper in phosphate buffered saline (PBS), using 10 ml of PBS every 10 x 106 cells and collect the volume of scraped cells in 50 ml plastic tubes.
- From this point keep cells cold at all time.
- Centrifuge for 10 min at 3,000 x g at 4 °C to pellet cells. Discard the supernatant.
- Wash the cell pellet with 20 ml of cold PBS, then centrifuge for 10 min at 3,000 x g at 4 °C and discard the supernatant.
- Suspend the cells in 10 ml of homogenization solution and disrupt cytoplasmic membranes and cytoplasm by homogenization with pestle and potter tissue grinder with a motor driven device: 5 strokes might be sufficient.
- Let the homogenized cells stand on ice for 10 min.
- At this point check whether the cytoplasmic membranes are broken up and the nuclei are intact and cytoplasm free. Mount approximately 10-20 μl of homogenized cells on a slide and observe with an optical microscope (Figure 1). If necessary you can repeat the homogenization step without exceeding the 10 strokes.
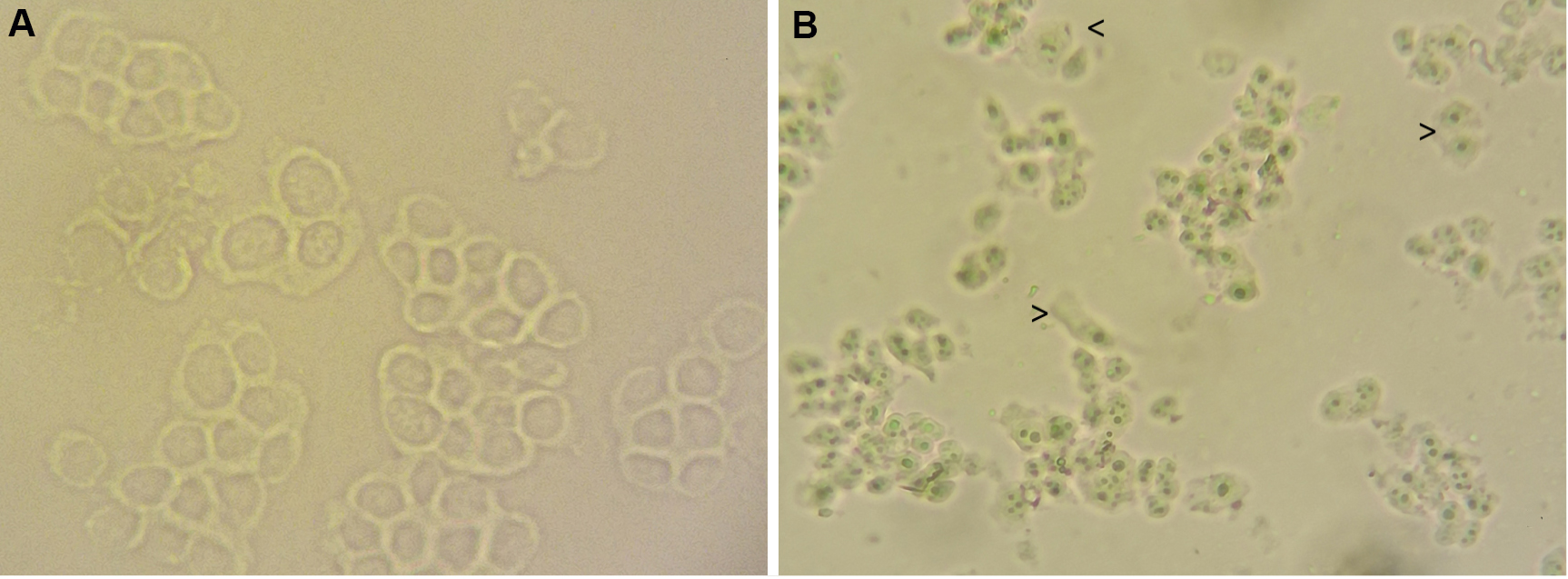
Figure 1. Microscopic images showing PLC whole and homogenized cells. A. PBS washed cells; B. Homogenized cell. By comparing with figure A, in B it is observable the breakage of cytoplasmic membranes and the loss of cell morphology. The nuclei are evident and intact. Arrowheads indicate cytoplasmic debris around nuclei, these residues can be removed by washing. A Zeiss Primovert inverted microscope was used. 200x magnification. - Transfer the homogenized sample to a tube and centrifuge for 10 min at 3,000 x g at 4 °C to pellet nuclei and discard any cytoplasmic residues in the supernatant. Wash the nuclei by adding 5 ml of washing solution and centrifuge for 10 min at 3,000 x g at 4 °C. Remove supernatant completely.
- Add 1 ml of suspension solution, mix by pipetting, take 0.1 ml of suspended nuclei and transfer it into another tube to extract and quantify hydrolyzed DNA. Use the remaining volume to assay in vitro RNA polymerase I activity.
- Grow the desired cell line in the required culture medium on cell culture flasks until reaching about 50 x 106 cells at least.
- In vitro assessment of RNA polymerase I activity
A high ionic strength medium in presence of α-amanitin is used to evaluate RNA polymerase I. In a high ionic strength medium both RNA polymerase I and II are active, however, RNA polymerase II is inhibited by α-amanitin (Novello and Stirpe, 1970).
The measurement of each sample is carried out in triplicate accompanied by two blanks.- Prepare 10 ml glass tubes, each tube must contain a final volume of 500 μl.
Add in the following order:
100 μl of 5x high ionic strength buffer
90 μl of nucleoside triphosphate mixture
1 μl of 1 mg/ml of α-amanitin solution
MilliQ H2O to 400 μl
100 μl of nuclei
Note: Add nuclei to the tubes as the last element, vortex very fast and start reaction time immediately. - Incubate all the samples for 10 min at 37 °C in a water bath with shaking at 100 rpm.
- After 10 min, stop the reaction using 5 ml of 10% (w/w) TCA and putting tubes on ice. Keep tubes on ice until the filtration step.
Notes: - To prepare blank samples, add nuclei to the tubes and immediately block the reaction with 5 ml of 10% (w/w) trichloroacetic acid (TCA), then incubate for 10 min at 37 °C and after that put tubes on ice.
- Whereas RNA polymerase I activity accounts for at least 60% of the cell transcriptional activity, RNA polymerase III contributes to a very small percentage of the cell transcriptional activity and this assay is not optimal to discriminate its activity, so it is not taken into account. To completely exclude RNA polymerase III activity an α-amanitin dose ten times more concentrated than the one which inhibits RNA polymerase II is required (at least a final concentration of 10 μg/ml).
- Using a high ionic strength medium it is possible to measure both RNA polymerase I and II: RNA polymerase II activity may be obtained from the difference between the measurements in the absence and in the presence of α-amanitin.
- It is also possible to discriminate and evaluate the RNA polymerase I activity using a low ionic strength reaction mixture (see Recipe 15), which is optimal for RNA polymerase I enzyme, but not for RNA polymerase II.
- Prepare 10 ml glass tubes, each tube must contain a final volume of 500 μl.
- Newly synthesized rRNA collection and measurement
TCA precipitates nucleic acid molecules, including newly synthesized 3H-labelled rRNA, which can be collected on a glass-fiber filter and measured in a liquid scintillation counter.- To prepare for a vacuum filtration, gather together a 2,000 ml filter flask and a vacuum glass filter holder for 25 mm disc filters, connect the side arm of filter flask to a vacuum pump using vacuum tubing. Assemble the apparatus placing a wet glass microfiber filter disc on the base (Figure 2). The filter discs are wetted with 5% (w/w) TCA.
- Pour a sample in the funnel, turn the vacuum pump on until the entire sample has passed through the filter and funnel base. Rinse the filter with 5% (w/w) TCA, 10 ml each wash for 5 washings.
Note: The collected liquid in the filter flask contains most of the radioactivity. - Turn the vacuum pump off, remove the funnel and using forceps place the filter in a scintillation plastic vial containing 10 ml of scintillation liquid.
- Repeat steps C2 and C3 for each sample tube.
- Shake the vials vigorously and put them in a liquid scintillation counter to measure the radioactivity of the samples.

Figure 2. Assembly of the filtration system. A and B. Apparatus components: filter flask (1), glass microfiber filter discs (2), funnel (3), glass support base (4) inserted in a perforated stopper (5), forceps (6) and clamp (7). C. The stopper with glass support base is inserted in the flask opening. TCA wet glass microfiber filter is placed on the base. D. The funnel is put on the filter and stopped with the clamp. The side arm of the filter flask is connected to a vacuum pump by a vacuum tubing.
- To prepare for a vacuum filtration, gather together a 2,000 ml filter flask and a vacuum glass filter holder for 25 mm disc filters, connect the side arm of filter flask to a vacuum pump using vacuum tubing. Assemble the apparatus placing a wet glass microfiber filter disc on the base (Figure 2). The filter discs are wetted with 5% (w/w) TCA.
- Nuclear DNA extraction and quantification
The following procedure is based on what reported by Munro and Fleck (1966).- Use 0.1 ml of suspended nuclei to extract and quantify hydrolyzed DNA.
- Collect 0.1 ml of nuclei and transfer it into a 10 ml glass tube.
- Add 0.5 ml of 0.6 N perchloric acid (PCA), vortex the sample a few seconds and centrifuge for 10 min at 3,000 x g at 4 °C. Remove and discard the supernatant containing single nucleotides and oligonucleotides, not included in DNA and RNA molecules that are soluble in weakly acid solution. Maintain pellet containing nuclear RNA molecules and DNA.
- Add 0.5 ml of 0.3 N potassium hydroxide (KOH) to pellet, vortex the sample and incubate for 30 min at 37 °C in a water bath with shaking to hydrolyze RNA.
Note: In alkaline environment RNA is susceptible to base-catalyzed hydrolysis while DNA is chemically stable. - Add 50 μl of 6 N PCA to solubilize ribonucleotides and precipitate macromolecules that are still present. Centrifuge for 10 min at 3,000 x g at 4 °C, discard supernatant containing hydrolyzed RNA and retain pellet containing DNA.
- Add to pellet 0.5 ml of 7% (w/w) PCA, vortex the sample to suspend and incubate for 15 min at 70 °C in a water bath with shaking to hydrolyze DNA into deoxynucleotides.
Note: Acid solution and heating liberate deoxynucleotides, which are recoverable in supernatant after centrifugation. - Centrifuge for 10 min at 3,000 x g at 4 °C. Transfer the supernatant containing hydrolyzed DNA to a 1.5 ml tube.
- Use spectrophotometric measurements at the 260 nm wavelengths to quantitate the amount of hydrolyzed DNA.
- DNA concentration is calculated multiplying absorbance at 260 nm, normalized to a 10 mm path length, by a factor of 20:
DNA (μg/ml) = (OD260nm/NF) x 20
Where,
OD260nm (Optical Density): absorbance at the indicated wavelength,
NF (Normalization Factor): factor to normalize to a 10 mm path length, for example, if path length is 1 mm the normalization factor is 10,
Factor 20: 1 OD at 260 nm corresponds to approximately 20 μg/ml for oligonucleotides.
- Use 0.1 ml of suspended nuclei to extract and quantify hydrolyzed DNA.
Data analysis
RNA polymerase I activity is evaluated by the measurement of radioactivity, expressed in disintegrations per minute (dpm), incorporated into newly synthesized rRNA. Each cell sample is evaluated in triplicate.
- Calculate the dpm mean of the blanks.
- Subtract the blank mean from each replicated sample.
- Divide the results by the μg of DNA previously quantified and calculate the mean and the standard deviation. The mean is expressed in dpm/μg DNA.
Assuming that the DNA quantity is directly proportional to the number of nuclei, the result represents the quantity of 3H labelled rRNA newly synthesized by RNA polymerase I per nucleus.
Representative data
An example of RNA polymerase I activity measurement is described below. The representative data were obtained measuring the enzymatic activity in two human cancer cell lines, HCT11 and LoVo. These cell lines are characterized by different level of rRNA synthesis as reported in Scala et al. (2016) and have different cell cycle length.
- Figure 3 shows the output of radioactivity measurements by the liquid scintillation counter instrument.
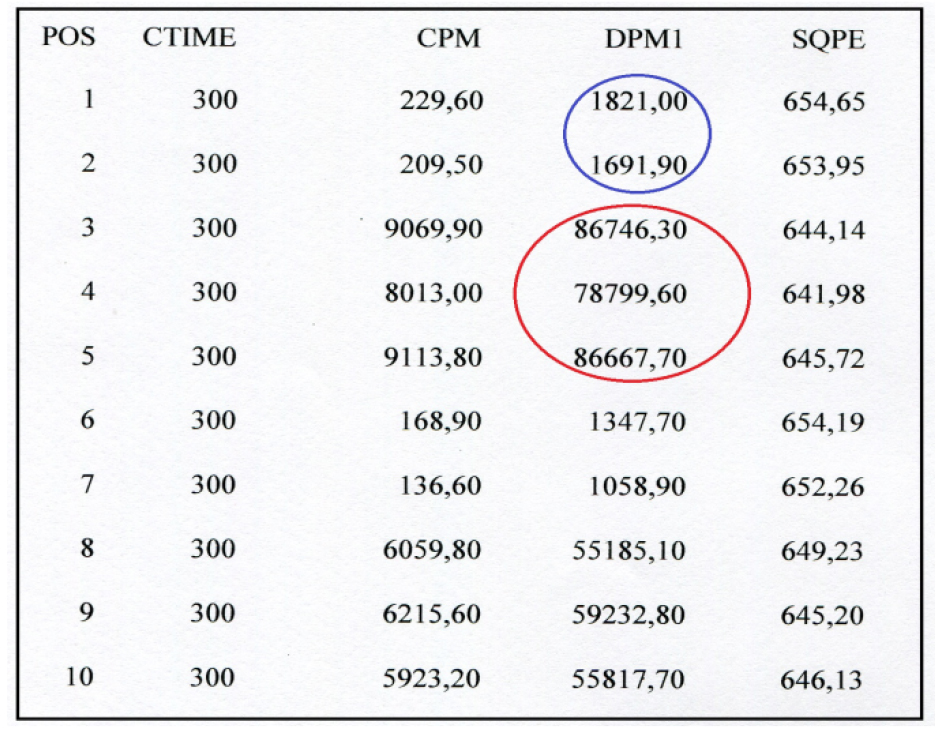
Figure 3. Picture of the radioactivity values obtained by reading the samples in the liquid scintillation counter (Guardian, Wallac, PerkinElmer). Radioactivity values are expressed in dpm. 1-5: HCT116 samples; 6-10 Lovo samples. Blue circle encloses two blanks, whereas red circle corresponds to the radioactivity measures of 3H uridine 5’ triphosphate amount incorporated in ex novo synthesized rRNA molecules by RNA polymerase I. The instrument provides both disintegrations per minute (dpm) and counts per minute (cpm); counts per minute are the counts received by an instrument from the source. - In Table 1 the data analysis is shown step by step. Once the radioactivity measures were obtained, the dpm means of the blanks were calculated and subtracted from each respective replicate. The results were divided by the μg of DNA present in 0.1 ml of nuclei. Then, the mean and the standard deviation of the blank subtracted dpm values normalized to 1 μg of DNA were calculated. All data were processed using Microsoft Excel program.
Table 1. Data analysis step by step. The table reports the data analysed in a worksheet of Excel program (Microsoft Excel). All data processed are in bold. The means of radioactivity values normalized to 1 μg of DNA are in red. The DNA content in 0.1 ml of nuclei suspension was calculated as indicated in Nuclear DNA extraction and quantification paragraph.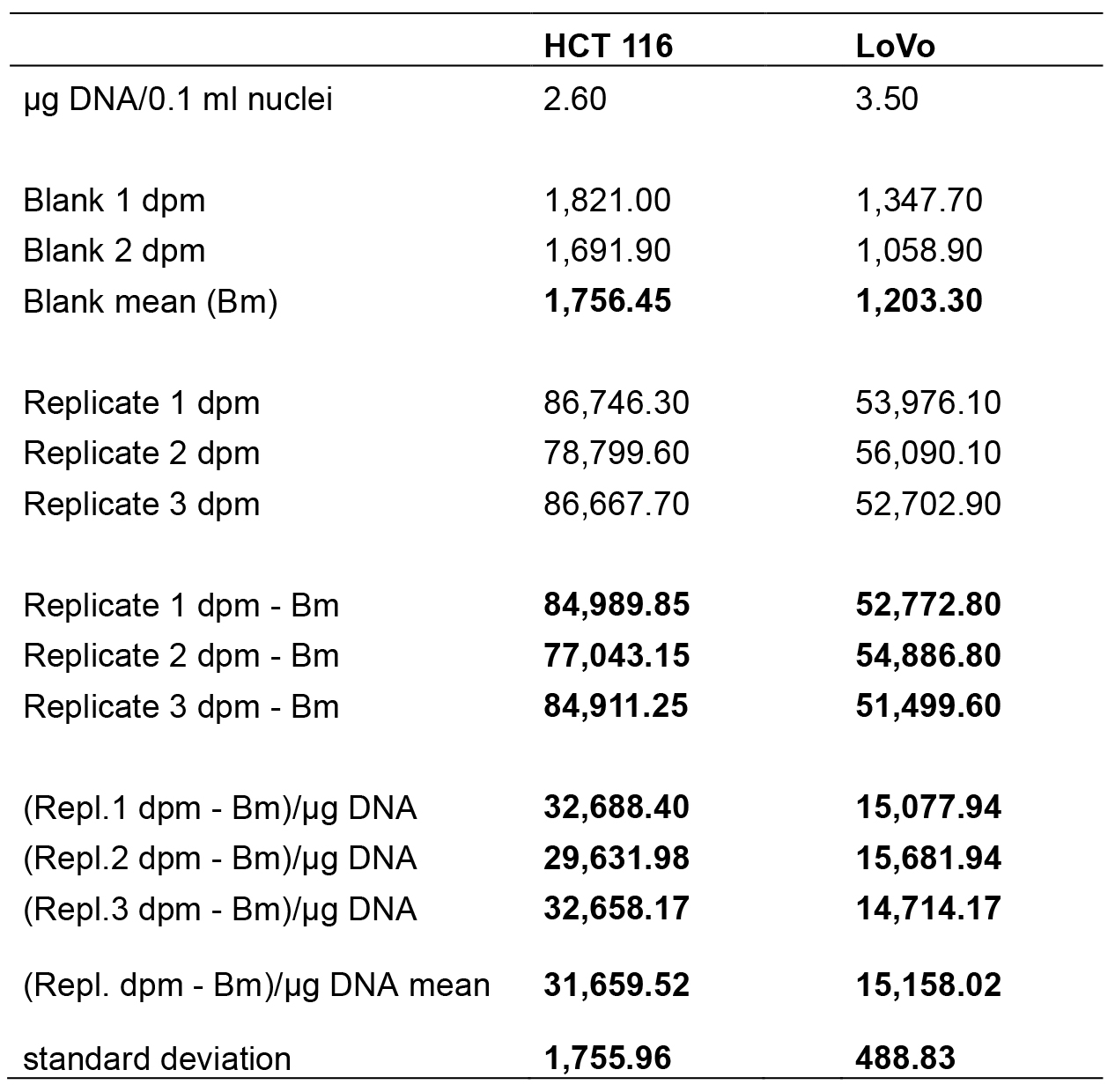
- In Figure 4 the final results are reported in a histogram, which makes clearer the different quantity of 3H labelled rRNA newly synthesized by RNA polymerase I in the two cancer cell lines.
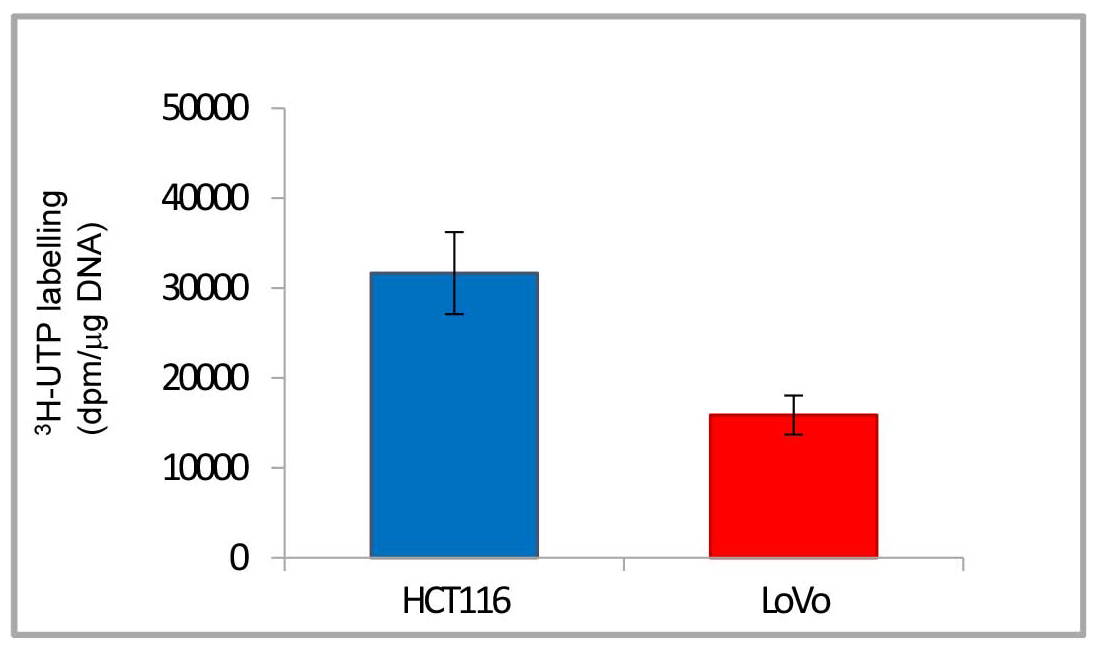
Figure 4. Histogram of representative data from Table 1. The histogram shows the final results: 3H-uridine 5’ triphosphate incorporated in rRNA newly synthesized by RNA polymerase I normalized to 1 μg of DNA (dpm/μg DNA). Data are shown as means ± standard deviation.
Recipes
- PBS (pH 7.3)
8.00 g NaCl
0.20 g KCl
1.44 g Na2HPO4
0.24 g KH2PO4
Dissolve in 800 ml of ddH2O
Adjust pH to 7.3 with HCl and add ddH2O to a final volume of 1 L
Final concentration: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4,1.8 mM KH2PO4 - 1 M Tris-HCl solution (pH 7.4 or pH 8.0)
12.11 g of Tris base
Dissolve in 80 ml of MilliQ H2O
Adjust pH to 7.4 (or pH 8.0) with the appropriate volume of concentrated HCl
Add MilliQ H2O to a final volume of 100 ml - 3 M ammonium sulfate
39.64 g of ammonium sulfate
Dissolve in 80 ml of MilliQ H2O
Add MilliQ H2O to a final volume of 100 ml - Homogenization-solution
8.56 g of sucrose
Dissolve in 50 ml of ddH2O
Add 1 ml of 1 M Tris-HCl pH 7.4 (see Recipe 2), 1 ml of 1 M MgCl2 and 0.5 g of Triton X-100
Add ddH2O to a final volume of 100 ml
Final concentration: 0.25 M sucrose, 10 mM Tris-HCl pH 7.4, 10 mM MgCl2, 0.5% Triton X-100 - Washing solution
Note: This solution is the same as homogenization solution without detergent.
8.56 g of sucrose
Dissolve in 50 ml of ddH2O
Add 1 ml of 1 M Tris-HCl pH 7.4, 1 ml of 1 M MgCl2
Add ddH2O to a final volume of 100 ml - Suspension solution
4.28 g of sucrose
Dissolve in 40 ml of ddH2O
Add 50 μl of 1 M MgCl2 and reach 50 ml of final volume with ddH2O
Final concentration: 0.25 M sucrose, 1 mM MgCl2 - High ionic strength 5x reaction mixture
2.5 ml 1 M Tris-HCl pH 8.0 (see Recipe 2)
0.1 ml 1 M MnCl2 (see Reagent 18)
2.0 ml 3 M ammonium sulfate (see Recipe 3)
0.4 ml MilliQ H2O
Final concentration in enzyme assay: 0.1 M Tris-HCl (pH 8.0), 4 mM MnCl2, 0.24 M ammonium sulfate - Nucleoside triphosphate mixture
Mix non-radioactive nucleoside triphosphate solution with radioactive solution in the proportions shown below.- Non-radioactive solution:
14.4 mg of GTP
12 mg of CTP
13.5 mg of ATP
0.48 mg of UTP
Dissolve in 2.5 ml of MilliQ H2O - Radioactive solution:
50 μCi of 3H-UTP diluted in
0.5 ml of MilliQ H2O
- Non-radioactive solution:
- α-amanitin reconstitution
1 mg/ml aqueous solution of α-amanitin
Store it at -20 °C
Note: You may store for a long time. It does not degrade with repeated freeze-thaw cycles. - 10% (w/w) TCA
Dilute 10 g of concentrated TCA (≥ 99% w/w) in 100 g of ddH2O - 0.6 N PCA
Dilute 2.9 ml of 65% (w/w) perchloric acid in 50 ml of ddH2O - 0.3 N KOH
Dissolve 0.842 g of KOH in 50 ml of ddH2O - 6 N PCA
Dilute 29 ml of 65% (w/w) PCA in 50 ml of ddH2O - 7% (w/w) PCA
Dilute 5.39 g of 65% (w/w) PCA in 50 g of ddH2O - 0.5 M NaF
Dissolve 0.3 g of NaF in 10 ml of MilliQ H2O - Low ionic strength 5x reaction mixture
2.5 ml 1 M Tris-HCl (pH 8.0) (see Recipe 2)
0.1 ml 1 M MgCl2 (see Reagent 16)
18 μl 2-mercaptoethanol (14 M)
0.3 ml 0.5 M NaF (see Recipe 15)
2.082 ml MilliQ H2O
Final concentration in enzyme assay: 0.1 M Tris-HCl (pH 8.0), 4 mM MgCl2, 50 μM 2-mercaptoethanol, 6 mM NaF
Note: Using low ionic strength medium the α-amanitin is not necessary and it is recommended a reduced volume of nucleoside triphosphate mixture (30 μl) because the blanks may be too high.
Acknowledgments
This protocol was adapted from the research article of Novello and Stirpe (1970). Work was supported by the Roberto and Cornelia Pallotti’s Legacy for Cancer Research. The author thanks Prof. Massimo Derenzini for the opportunity to describe this protocol and for help and suggestions. The author is also thankful to Dr. Christine M. Betts for language revision of the manuscript. The author declares no conflicts of interest.
References
- Derenzini, M., Montanaro, L., Chilla, A., Tosti, E., Vici, M., Barbieri, S., Govoni, M., Mazzini, G. and Trere, D. (2005). Key role of the achievement of an appropriate ribosomal RNA complement for G1-S phase transition in H4-II-E-C3 rat hepatoma cells. J Cell Physiol 202(2): 483-491.
- Donati, G., Bertoni, S., Brighenti, E., Vici, M., Treré, D., Volarevic, S., Montanaro, L. and Derenzini, M. (2011). The balance between rRNA and ribosomal protein synthesis up- and downregulates the tumour suppressor p53 in mammalian cells. Oncogene 30: 3274-3288.
- Montanaro, L., Trere, D. and Derenzini, M. (2013). The emerging role of RNA polymerase I transcription machinery in human malignancy: a clinical perspective. Onco Targets Ther 6: 909-916.
- Munro, H. N. and Fleck, A. (1966). Recent developments in the measurement of nucleic acids in biological materials. A supplementary review. Analyst 91(79): 78-88.
- Novello, F. and Stirpe, F. (1970). Simultaneous assay of RNA polymerase I and II in nuclei isolated from resting and growing rat liver with the use of alpha-amanitin. FEBS Lett 8(1): 57-60.
- Scala, F., Brighenti, E., Govoni, M., Imbrogno, E., Fornari, F., Treré, D., Montanaro, L. and Derenzini, M. (2016). Direct relationship between the level of p53 stabilization induced by rRNA synthesis-inhibiting drugs and the cell ribosome biogenesis rate. Oncogene 35(8): 977-989.Let the homogenized cells stand
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Govoni, M. (2017). In vitro Assessment of RNA Polymerase I Activity. Bio-protocol 7(3): e2120. DOI: 10.21769/BioProtoc.2120.
Category
Cancer Biology > General technique > Biochemical assays > Protein analysis
Biochemistry > Protein > Activity
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.