- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Measuring Procaspase-8 and -10 Processing upon Apoptosis Induction


Published: Vol 7, Iss 1, Jan 5, 2017 DOI: 10.21769/BioProtoc.2081 Views: 10546
Reviewed by: Emilie BesnardAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Apr 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols

Abstract
Apoptosis or programmed cell death is important for multicellular organisms to keep cell homeostasis and for the clearance of mutated or infected cells. Apoptosis can be induced by intrinsic or extrinsic stimuli. The first event in extrinsic apoptosis is the formation of the Death-Inducing Signalling Complex (DISC), where the initiator caspases-8 and -10 are fully activated by several proteolytic cleavage steps and induce the caspase cascade leading to apoptotic cell death. Analysing the processing of procaspases-8 and -10 by Western blot is a commonly used method to study the induction of apoptosis by death receptor stimulation. To analyse procaspase-8 and -10 cleavage, cells are stimulated with a death ligand for different time intervals, lysed and subjected to Western blot analysis using anti-caspase-8 and anti-caspase-10 antibodies. This allows monitoring the caspase cleavage products and thereby induction of apoptosis.
Keywords: Caspase-8Background
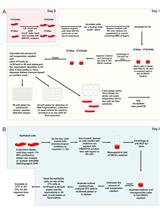
Caspases are proteases that are produced as inactive zymogens and are activated by proteolytic cleavage (Degterev et al., 2003). The activation of the caspase cascade is the most important event during apoptotic cell death, which induces the typical biochemical and morphological changes of the apoptotic cell. In contrast to inactive executioner procaspases, the initiator procaspases 8/9/10 have restricted proteolytic activity and become fully activated in high molecular weight complexes (Lavrik et al., 2005). Procaspase-9 is activated at the platform termed apoptosome during intrinsic apoptosis, while stimulation of the death receptors TNF-R1 (Tumor Necrosis Factor Receptor 1), CD95/Fas (Cluster of Differentiation 95), TRAIL-R1 (Tumor Necrosis Factor Related Apoptosis Inducing Ligand Receptor 1), TRAIL-R2 (Tumor Necrosis Factor Related Apoptosis Inducing Ligand Receptor 2) leads to the recruitment of procaspases-8 and -10 with adapter proteins to form the Death-Inducing Signalling Complex (DISC) during extrinsic apoptosis (Schleich et al., 2012). Here, caspases-8 and -10 enter close proximity and perform several intra- and inter-molecular cleavages. This processing results in the release of a small and a large subunit from the prodomain. These form the active heterotetramers p182p102 and p172p122 that triggers the caspase cascade leading to the demolition of the cell (see Figures 1A and 1B).
To verify apoptosis induction, it is important to show the activation of caspases. Checking the activation of the initiator procaspases 8 and 10, accompanied by their cleavage, gives main evidence for the induction of the extrinsic apoptosis via death receptors. The kinetics of procaspases-8 and -10 processing can be analysed by monitoring its cleavage steps by Western blot (see Figure 1C) and give information about the induction of apoptotic cell death after death receptor stimulation (Schleich et al., 2016). Depending on the part of the caspase that is recognized by the antibody, the intermediate products of the caspase containing this particular part can be analysed by western blot. Healthy, unstimulated cells only contain the unprocessed procaspases-8 and -10 (p55/53 and p59/55, respectively), but after stimulation the amount of procaspase decreases and the intermediate cleavage products (caspase-8 p43/41 or caspase-10 p47/43) and active subunits (caspase-8 p18) are enriched and can be detected by Western blot (Figure 1C). By performing time-dependent analysis, it is possible to follow the course of caspase cleavage including the enrichment of the cleaved forms (Schleich et al., 2016). This also allows to compare the time-dependent cleavage of caspases between several conditions.
To ensure getting the complete information on procaspase-8/-10 processing we have developed the protocol presented below. In contrast to a number of other protocols for Western blot analysis of procaspase-8/-10 processing, here we do not follow only a part of the Western blot of a specific molecular weight, e.g., by cutting the membrane, or use antibodies specific only to the active forms of the caspases. In this way, we can follow simultaneously several cleavage products of procaspases-8 and -10: proform, intermediate and final cleavage products. Only this way of measuring procaspase-8/-10 processing allows to escape from misjudgement on the efficiency of procaspase cleavage, e.g., in some studies only the proform of procaspases is followed and in case of a low stimulation strength and a weak caspase processing, the small differences in the decrease of the proform are not detected and might be misleading. Furthermore, another very important feature of our protocol is performing the measurement over different time intervals, which is also often neglected, and thus, the results might be misleading due to missing the key time points of processing, for example the appearance of active caspases, that are degraded fast. To the later point, there is a cell type and stimulation strength specificity with respect to the timing of procaspase-8/-10 processing and the corresponding time intervals have to be carefully selected in each particular case.
Figure 1. Kinetics of procaspase-8 and procaspase-10 processing. A. Scheme of caspase-8 processing. The cleavage at D216, D374 and D384 results in the release of the active subunits (p18 and p10). The point marks the region of the caspase that is recognized by the antibody (epitope). B. Scheme of caspase-10 processing. The cleavage at D219, D372 results in the release of the active subunits (p23/p17 and p12). The point marks the region of the caspase that is recognized by the antibody (epitope). C. Western blot: Hela-CD95 cells (Neumann et al., 2010) were stimulated with 250 ng/ml CD95L for the indicated periods of time. Samples were lysed, subjected to SDS-PAGE and analysed by Western blot for caspase-8 and caspase-10. Western blot for actin was used as loading control.
Materials and Reagents
- 6-well plates (with a surface suitable for your cells, e.g., SARSTEDT, catalog number: 83.3920 )
- Microcentrifuge tubes (e.g., VWR, catalog number: 89202.684 )
- Cuvettes (e.g., SARSTEDT, model: 67.742 )
- Blotting membrane and Whatman paper (e.g., Trans-Blot Turbo Transfer Pack Mini incl. 0.2 µm nitrocellulose and transfer buffer, Bio-Rad Laboratories, catalog number: 170-4158 )
- Human cells expressing caspase-8 and caspase-10, here: HeLa cells stably overexpressing CD95 (Neumann et al., 2010; DKMZ, catalog number: ACC 57 )
- Suitable medium for your cells (e.g., DMEM/Ham’s F12 including 10% fetal bovine serum and 1% penicillin/streptomycin)
- CD95L has been prepared as described in Fricker et al., 2010
- Bovine serum albumin (BSA) standard 2 mg/ml (Bio-Rad Laboratories, catalog number: 5000260 )
- SDS-poly acrylamide gel (e.g., Mini-PROTEAN TGX stain-free precast gels, 12%, Bio-Rad Laboratories, catalog number: 456-8046 )
- Protein assay dye reagent concentrate (Bio-Rad Laboratories, catalog number: 500-0006 )
- Protein standard (e.g., Precision plus Protein All Blue standards, Bio-Rad Laboratories, catalog number: 1610373 )
- Anti-caspase-8 antibody, clone C15 (final concentration 0.5-2 µg/ml, a kind gift of P. Krammer, DKFZ Heidelberg)
- Anti-caspase-10 antibody, clone 4C1 (dilution 1:1,000, final concentration 1 µg/ml, MEDICAL & BIOLOGICAL LABORATORIES, catalog number: MBL-M059-3 )
- Anti-actin antibody (dilution: 1:4,000, final concentration 0.125-0.2 µg/ml, Sigma-Aldrich, catalog number: A2103 )
- HRP-coupled isotype specific secondary antibodies (e.g., Santa Cruz Biotechnology, catalog numbers: sc-2060 , sc-2004 , sc-2062 )
- HRP-Substrate for Enhanced Luminescence, (e.g., Luminata Forte Western HRP Substrate, EMD Millipore, catalog number: WBLUF0500 )
- 10x PBS (Biochrom, catalog number: L1835 )
- cOmpleteTM protease inhibitor cocktail (PIC) (Roche Diagnostics, catalog number: 11 836 145 001 )
- Tris (AppliChem, catalog number: A2264 )
- Natrium chloride (NaCl) (Carl Roth, catalog number: P029.3 )
- EDTA (Carl Roth, catalog number: CN06.2 )
- Glycerol (Carl Roth, catalog number: 3783.1 )
- Triton (Carl Roth, catalog number: 3051.4 )
- Tween-20 (AppliChem, catalog number: A1389 )
- Milk powder (Carl Roth, catalog number: T145.5 )
- Sodium azide (Carl Roth, catalog number: K305.1 )
- β-mercaptoethanol (Carl Roth, catalog number: 4227.2 ).
- 10 x Tris/Glycine/SDS (Bio-Rad Laboratories, catalog number: 161-0723 )
- 4x Laemmli sample buffer (Bio-Rad Laboratories, catalog number: 161-0747 )
- 1x PBS (see Recipes)
- Protease inhibitor cocktail (PIC, see Recipes)
- Lysis buffer (see Recipes)
- 1 x Tris/Glycine/SDS (see Recipes)
- PBS-T (see Recipes)
- Blocking buffer (see Recipes)
- Primary antibody dilution (see Recipes)
- 4x Laemmli sample buffer (reducing, see Recipes)
Equipment
- CO2-incubator (e.g., Thermo Fisher Scientific, Thermo ScientificTM, model: BBD 6220 )
- Cell scraper (e.g., Orange Scientific, catalog number: 4460600N )
- Centrifuge (Eppendorf, model: 5418R )
- Photometer (Bio-Rad Laboratories, model: SmartSpec Plus Spectrophotometer )
- Electrophoresis chamber and power supply (Bio-Rad Laboratories, model: Mini-PROTEAN Tetra cell and PowerPac HC )
- Transfer system (Bio-Rad Laboratories, model: Trans-Blot Turbo Transfer System )
- Imager (Bio-Rad Laboratories, model: ChemiDoc XRS+ Imaging System )
Software
- Image Lab (Bio-Rad Laboratories)
Procedure
- Cell lysis
- Plate 2.5 x 105 HeLa-cells per well on 6-wells in 2 ml medium (see Materials and Reagents 6) and incubate overnight at 37 °C, 5% CO2.
- Next day control cells, aspirate medium and replace with fresh medium. Stimulate cell with apoptosis inducing agent (here 250 ng/ml CD95L) in a time-dependent manner (here: 20, 40, 60, 90, 120 min). Stimulation dose and time is cell line dependent and have to be tested for each cell line. Stimulation should be performed in a way that one 6-well plate can be harvested at once.
- Incubate the cells at 37 °C, 5% CO2.
- Add cold PBS to the cells (do not aspirate media), harvest them by scraping and transfer medium including cells into a new tube (everything on ice). Be careful to transfer all the cells into the new tube.
- Centrifuge at 4 °C, 500 x g for 5 min.
- Discard supernatant, but not the cell pellet!
- Add 1 ml of cold PBS.
- Centrifuge at 4 °C, 500 x g for 5 min.
- Aspirate and discard supernatant.
- Store the pellet at -20 °C or continue with cell lysis by resuspending the cell pellet directly in 30 µl ice-cold lysis buffer (see Recipe 3), be careful to resuspend it completely.
- Incubate on ice for 30 min, vortex 2-3 times.
- Centrifuge at 4 °C, 16,400 x g for 15 min.
- Transfer the supernatant (= lysate) into a new tube, discard cell pellet. The supernatant should have a clear, yellowish colour.
- Measurement of protein concentration
- Measure protein concentration by mixing 2 µl of lysate with 1,000 µl Bio-Rad assay dye reagent (see Recipe 4) in a cuvette.
- Prepare a BSA-standard with dilutions according to manufacturer’s instructions.
- Mix 2 µl lysis buffer with 1,000 µl Bio-Rad solution and use it as blank.
- Incubate at room temperature for 5 min. The blank should have a brown-red colour, while the samples with lysates should turn into blue.
- Measure protein concentration for samples and BSA-standard at 595 nm with Bio-Rad SmartSpec plus or other spectrophotometer. Follow instructions of spectrophotometer for protein standard. Be careful to measure within the linear range and to dilute samples with too high protein concentrations. The expected protein concentration is between 3 and 5 mg/ml.
- SDS-PAGE and blot transfer
- Prepare 25 µg of protein lysate with lysis buffer and 4x Laemmli sample buffer (reducing, see Recipe 9) to a final volume of 15 µl (e.g., mix 5 µl of a 5 µg/µl lysate with 6.25 µl lysis buffer and 3.75 µl sample buffer). If the protein concentration is too low you can use gels with higher loading volume or decrease protein amount to 20 µg.
- Heat the samples at 95 °C for 5 min.
- Spin down shortly to avoid steam on the lid.
- Prepare a chamber with gel and 1x Tris/Glycine/SDS (Sodium dodecyl sulfate) buffer (see Recipe 5).
- Load samples on 12% Bio-Rad pre-cast stain-free gels. Be careful to load the complete sample into the well without leakage and spillover. Use of 25 µl Hamilton syringe or long pipette tips makes loading on a gel easier. Put the end of the tip or the syringe to the bottom of the well and very slowly push the sample into the well. Then carefully remove the Hamilton syringe or the tip from the well without spillover of the sample. When using Hamilton syringe rinse it three times in-between loading the next sample.
- Load 2 µl of all blue protein standard.
- Run the gel with 150 V until the protein dye passes the bottom of the gel. Check the progress from time to time.
- Place gel in Tris/Glycine/SDS buffer.
- If you use Bio-Rad stain-free gels, activate the dye in the gel with one minute UV light, take a picture of the gel and check the separation of the proteins. It is not necessary to use Bio-Rad stain-free gel system. The system is based on the implementation of chemical compounds that react with tryptophan under UV light resulting in fluorescence. The fluorophore is visible on the membrane after blotting.
- Prepare a sandwich consisting of Whatman paper sheets, a membrane (PVDF or Nitrocellulose) = Blot, the gel and Whatman paper sheets in presence of a transfer buffer. Be careful to place the sandwich in the blotting system oriented to the cathode, so that the proteins will be transferred from the gel to the membrane, not into the Whatman paper. Blot with Transblot Turbo transfer system according to instruction with 2.5 A, 25 V for 9 min. It is possible to use a different system for Western blot as well.
- Rinse blot with PBS-T (PBS-Tween, see Recipe 6) for a short time.
- If you are using Bio-Rad stain-free gels, take a picture of the blot and check blotting results for air bubbles and equal transfer.
- Membrane blocking and detection of caspase-10
- Incubate in blocking buffer (see Recipe 7) for 1 h shaking at room temperature. The blot should be covered completely with buffer.
- Rinse 3 x for 5 min with PBS-T, spill buffer into the dish, not directly onto the blot to avoid removing of proteins.
- Incubate with primary antibody dilution (see Recipe 8) overnight shaking at 4 °C. You can use the antibody solution several times, but store it a 4 °C and check for contamination before using it. When the antibody signal is fading, prepare a new antibody solution.
- Wash blot 4 x with PBS-T for 5 min, shaking at room temperature.
- Prepare a 1:10,000 dilution of secondary antibody in blocking buffer, approx. 20 ml/blot.
- Incubate the blot in the secondary antibody dilution for 60 min at room temperature while shaking.
- Wash blot 3 x with PBS-T for 10 min while shaking at room temperature.
- Rinse blot with 1 ml Luminata forte solution and wait a few seconds, the complete blot should be covered with the solution.
- Place blot in the imager machine and scan the blot.
- Wash blot 3 x in PBS-T for 5 min.
- Detection of caspase-8 and actin
- Repeat steps of D3-D10 for caspase-8 and actin antibody detection. The use of isotype specific secondary antibodies allows to detect caspase-8 after caspase-10 on the same membrane. Alternatively it is possible to use two separate blots for detection of caspase-8 and 10 (see Figure 1C).
Data analysis
Check the quality of your blot, the bands should appear sharp without any smear or air bubbles. By adding a positive control to your gel, it will be possible to check the quality of the blot and the detection of the caspase fragments. For example, the caspase-8 cleavage products p43/41 and p18 should be visible (Figure 1C). A possible positive control could be lysate from apoptotic dying cells (e.g., HeLa-CD95 cells stimulated for 120 min with 250 ng/ml CD95L). Next to this qualitative analysis of the Western blot data, they can be quantified, too. This requires a high-quality blot and antibody signals within the detection range (no overexposure). There are several approaches to perform the quantitative analysis, one can be found here: Schleich et al., 2016.
Notes
- Mice do not express caspase-10, they only express caspase-8.
- The optimal time point and concentration for detection of caspase-8 and -10 activation have to be optimized for each cell line and stimulus.
- You can use the primary antibody dilution several times until it is contaminated or efficiency of detection is fading. Store at 4 °C.
- It is possible to use TBS-T instead of PBS-T for blot washing.
- Washing buffers can be prepared with deionized water instead of MilliQ water.
Recipes
- 1x PBS
For 1 L add 100 ml 10x PBS to 900 ml MilliQ water - Protease inhibitor cocktail (PIC)
1 tablet of cOmpleteTM protease inhibitor cocktail
Dilute in 2 ml of MilliQ water to receive 25x PIC solution
Store in aliquots at -20 °C - Lysis buffer (store at 4 °C)
20 mM Tris (pH 7.4)
137 mM NaCl
2 mM EDTA
10% glycerol
1% Triton
Add 4% (v/v) 25x PIC at the day of use (see Recipe 2) - Bio-Rad protein assay dye reagent (100 ml)
For 100 ml add 20 ml Bio-Rad protein assay dye reagent concentrate to 80 ml MilliQ water
Use up to 3 weeks and store at RT - 1x Tris/glycine/SDS
For 1 L add 100 ml 10 x Tris/glycine/SDS to 900 ml water - PBS-T
Add 2 ml Tween-20 to 2 L 1x PBS and mix - Blocking buffer
Mix 50 g milk powder in 1 L PBS-T until solved completely - Primary antibody dilution
Dilute antibody according to manufacturer’s instruction (see Materials and Reagents) in PBS-T Add sodium azide to a final concentration of 0.1% (w/v)
Store the antibody dilution at 4 °C - 4x Laemmli sample buffer (reducing)
Mix 900 µl of 4x Laemmli sample buffer with 100 µl β-mercaptoethanol according to the manufacturer’s instructions
Acknowledgments
The work has been supported by RSF 14-44-00011. This protocol was adapted from Schmidt et al., 2015; Pietkiewicz et al., 2015 and Schleich et al., 2016.
References
- Degterev, A., Boyce, M. and Yuan, J. (2003). A decade of caspases. Oncogene 22(53): 8543-8567.
- Fricker, N., Beaudouin, J., Richter, P., Eils, R., Krammer, P. H. and Lavrik, I. N. (2010). Model based dissection of CD95 signaling dynamics reveals both a pro- and anti-apoptotic role of c-FLIPL. J Cell Biol 190(3): 377-389.
- Lavrik, I. N., Golks, A. and Krammer, P. H. (2005). Caspases: pharmacological manipulation of cell death. J Clin Invest 115(10): 2665-2672.
- Neumann, L., Pforr, C., Beaudouin, J., Pappa, A., Fricker, N., Krammer, P. H., Lavrik, I. N. and Eils, R. (2010). Dynamics within the CD95 death-inducing signaling complex decide life and death of cells. Mol Syst Biol 6(1): 352.
- Pietkiewicz, S., Eils, R., Krammer, P. H., Giese, N. and Lavrik, I. N. (2015). Combinatorial treatment of CD95L and gemcitabine in pancreatic cancer cells induces apoptotic and RIP1-mediated necroptotic cell death network. Exp Cell Res 339(1): 1-9.
- Schleich, K., Buchbinder, J. H., Pietkiewicz, S., Kahne, T., Warnken, U., Ozturk, S., Schnolzer, M., Naumann, M., Krammer, P. H. and Lavrik, I. N. (2016). Molecular architecture of the DED chains at the DISC: regulation of procaspase-8 activation by short DED proteins c-FLIP and procaspase-8 prodomain. Cell Death Differ 23(4): 681-694
- Schleich, K., Warnken, U., Fricker, N., Ozturk, S., Richter, P., Kammerer, K., Schnolzer, M., Krammer, P. H. and Lavrik, I. N. (2012). Stoichiometry of the CD95 death-inducing signaling complex: experimental and modeling evidence for a death effector domain chain model. Mol Cell 47(2): 306-319.
- Schmidt, J. H., Pietkiewicz, S., Naumann, M. and Lavrik, I. N. (2015). Quantification of CD95-induced apoptosis and NF-κB activation at the single cell level. J Immunol Methods 423: 12-7.
Article Information
Copyright
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Pietkiewicz, S., Wolfe, C., Buchbinder, J. H. and Lavrik, I. N. (2017). Measuring Procaspase-8 and -10 Processing upon Apoptosis Induction. Bio-protocol 7(1): e2081. DOI: 10.21769/BioProtoc.2081.
Category
Cancer Biology > Cell death > Cell biology assays > Apoptosis
Immunology > Immune cell function > Cytotoxicity
Molecular Biology > Protein > Expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.