- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Isolation of Latex Bead Phagosomes from Dictyostelium for in vitro Functional Assays





Published: Vol 6, Iss 23, Dec 5, 2016 DOI: 10.21769/BioProtoc.2056 Views: 10422
Reviewed by: Kristopher MarjonGeneviève BallAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Feb 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
We describe a protocol to purify latex bead phagosomes (LBPs) from Dictyostelium cells. These can be later used for various in vitro functional assays. For instance, we use these LBPs to understand the microtubule motor-driven transport on in vitro polymerized microtubules. Phagosomes are allowed to mature for defined periods inside cells before extraction for in vitro motility. These assays allow us to probe how lipids on the phagosome membrane recruit and organize motors, and also measure the motion and force generation resulting from underlying lipid-motor interactions. This provides a unique opportunity to interrogate native-like organelles using biophysical and biochemical assays, and understand the role of motor proteins in phagosome maturation and pathogen clearance.
Keywords: Phagosome maturationBackground
In vitro reconstitution of biological processes is important to understand the molecular components and mechanisms underlying them. One such process is phagosome maturation, which is involved in degradation of pathogens taken up by macrophage cells of the immune system, and is also used as a process of nutrition in lower eukaryotes (Vieira et al., 2002). The transport of phagosomes on microtubules is intimately connected to their maturation (Blocker et al., 1997; Vieira et al., 2002). Notably, several intracellular pathogens disrupt phagosome transport to survive in a latent form inside cells (Harrison et al., 2004; Harrison and Grinstein, 2002; Rai et al., 2016; Sun et al., 2007). Therefore, reconstitution of phagosome transport might help to understand the strategy used by pathogens for immune evasion. Here, we describe a detailed protocol to prepare LBPs from cell extracts of the social amoeba Dictyostelium discoideum. This protocol has been adapted and modified from the work by Gotthard et al. (2006). Briefly, the cells are pulsed with latex beads and chased for different time durations to enable either early or late phagosome formation. Such phagosomes are buoyant in nature and float away from other endogenous vesicles when spun at high speeds. These phagosomes, collected along with the cytosol, show robust motion on in vitro polymerized microtubules. A detailed version of this protocol has also been published elsewhere (Barak et al., 2014). Using this method, we have recently shown cholesterol as a key regulator of phagosome transport and maturation (Rai et al., 2016). Furthermore, this assay has helped us to elucidate the mechanism of disruption of phagosome transport by lipophosphoglycan (LPG) from the parasite Leishmania donovani. A description of the phagosome extract preparation from Dictyostelium cells is detailed below. This protocol describes only the purification of LBPs. The in vitro motility assay has been described elsewhere (Barak et al., 2014).
Materials and Reagents
- Glass coverslip
- 1.5 ml microfuge tube
- Preassembled Acrodisc® syringe filters for lysing cells (5 μm pore size, Supor® membrane 32 mm diameter) (Pall, catalog number: 4650 )
- Syringes of 1-2 ml capacity for lysis
- 1 ml syringe with needle (26 G) for collection of LBPs
- Dictyostelium discoideum AX-2 strain cells (dictyBase, catalog number: DBS0238585 ) (see Note 1)
- HL-5 medium for cell culture: HL-5 medium with glucose (ForMediumTM, catalog number: HLG0102 ) prepared according to manufacturer’s specifications (see Note 2)
- Polystyrene beads: carboxylated polystyrene beads of 750 nm diameter (Polysciences, catalog number: 07759-15 ) (see Note 4)
- Penicillin-streptomycin (Penstrep) (10,000 μg/ml) (Thermo Fisher Scientific, GibcoTM, catalog number: 15140-122 )
- Protease inhibitor cocktail (cOmplete EDTA-free) (Roche Diagnostics, catalog number: 11836145001 )
- Liquid nitrogen for snap freezing
- Pepstatin A (MP Biomedical, catalog number: 2195368 )
- Methanol
- KH2PO4
- Na2HPO4
- Tris
- EGTA
- Sucrose
- DL-Dithiothreitol (DTT) (Sigma-Aldrich, catalog number: 43819 )
- Phenylmethanesulfonyl fluoride (PMSF) (Sigma-Aldrich, catalog number: 78830 )
- Benzamidine hydrochloride (Sigma-Aldrich, catalog number: 434760 )
- Sorensen’s buffer (see Recipes)
- Cell lysis buffer (see Recipes)
- Centrifugation cushion buffer (see Recipes)
Equipment
- Rotatory shaker
- Differential Interference Contrast (DIC) microscope (Nikon Instruments, model: TE2000U or similar)
- Cell culture microscope with 10x and 20x objective for observing and counting cells
- Water bath sonicator (Branson 1510MT ultrasonic cleaner, frequency 40 kHz, 10 min)
Note: This product has been discontinued. - Clinical centrifuge for pelleting cells
- Shaking incubator at 22 °C
- Autoclaved 500 ml conical flasks for shaking suspension culture
- Beckman Coulter centrifuge with JA-10 rotor
- Beckman Coulter table top ultracentrifuge with MLS-50 rotor
Procedure
Note: Steps mentioned in this protocol should be performed on ice in a walk-in cold room at 4 °C (unless stated otherwise). In case of unavailability of such a room, all steps should be performed strictly on ice.
- Culturing of Dictyostelium cells
Dictyostelium AX-2 cells are cultured in HL-5 suspension media with Penstrep (100 μg/ml working concentration) at 22 °C and 150 rpm in a shaking incubator (see Note 6). The optimal cell density for phagosome extract preparation is between 4-8 x 106 cells/ml. A 100 ml suspension culture (or 4-8 x 108 cells) is usually sufficient for one preparation (see Note 7). - Before each preparation a small aliquot of cells from the culture (50 µl) is put on a glass coverslip to observe motility of organelles inside cells under a 100x objective of a differential interference contrast (DIC) microscope. A video for the intracellular motility is shown (Video 1). Cells with poor intracellular motility and/or excess vacuoles are under stress, and should not be used. Vacuoles are easily observable as large membranous structures inside the cells (Figure 1B).
If cells appear healthy, before proceeding for the extraction procedure, it is required to perform the preparatory tasks as outlined in Note 3. Video 1. Intracellular motility of organelles in Dictyostelium. Cell suspension was placed on a clean glass coverslip, allowed to adhere to the surface for 3-5 min and then imaged under 100x objective of a DIC microscope. The movie was acquired at 30 frames/sec using a custom program written on LabVIEW and later reduced to 10 frames/sec. Movie runs in real-time. Organelles appear to move in a directed manner inside the cell. Robust in vivo motion of organelles (as observed in the video) is imperative for isolating highly motile LBPs.
Video 1. Intracellular motility of organelles in Dictyostelium. Cell suspension was placed on a clean glass coverslip, allowed to adhere to the surface for 3-5 min and then imaged under 100x objective of a DIC microscope. The movie was acquired at 30 frames/sec using a custom program written on LabVIEW and later reduced to 10 frames/sec. Movie runs in real-time. Organelles appear to move in a directed manner inside the cell. Robust in vivo motion of organelles (as observed in the video) is imperative for isolating highly motile LBPs.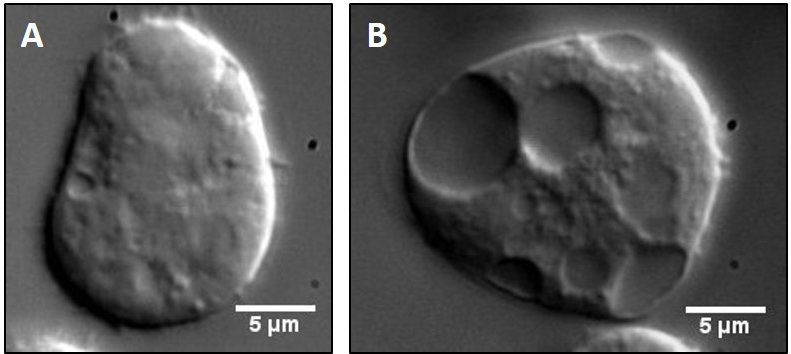
Figure 1. Comparison of healthy versus unhealthy Dictyostelium cells. Cell suspension from either an optimally dense (A) or highly dense (B) culture was placed on a clean glass coverslip, allowed to adhere to the surface for 3-5 min and then imaged under 100x objective of a DIC microscope. Stressed cells rarely adhere to the coverslip, appear spherical and show large vacuoles as opposed to healthy cells. Such cells also show highly diffusive intracellular motility and hence cannot be used for in vitro reconstitution experiments. - Bead preparation
The 750 nm bead stock solution mentioned in the materials section has a bead density of 1.08 x 1011 particles/ml. 200 µl of the bead stock solution is taken in a 1.5 ml microfuge tube and beads are pelleted by centrifuging at 10,000 x g for 5 min at 4 °C. The supernatant is discarded and the bead pellet is resuspended in 1 ml of HL-5 medium (see Note 8). This washing step is repeated once more and the final bead pellet is resuspended in 500 µl of Sorensen’s buffer. In order to avoid clumping of beads, they are sonicated in a sonicating water bath for 10 min and kept on ice until further use. - Cells are collected by centrifuging the suspension culture twice in a 50 ml Falcon tube at 900 x g for 3 min at room temperature. The cell pellet is immediately stored on ice and resuspended in 5 ml of ice-cold Sorensen’s buffer.
- Synchronization
The washed bead solution (500 µl) is added to the cells and the bead-cell suspension is incubated at 4 °C for 20 min with gentle shaking on a rotatory shaker (see Note 9). - Pulse
After synchronization, the bead-cell suspension is added to 100 ml of HL-5 medium kept in a 500 ml conical flask at 22 °C to initiate bead uptake. The incubation is done at 22 °C and 150 rpm in a shaking incubator. To isolate early phagosomes, a pulse duration of 5 min (see Note 10) is used. To isolate late phagosomes, the cells are pulsed with beads for 15 min. - To stop the pulse, the 100 ml cell suspension is directly added to 330 ml of ice-cold Sorensen’s buffer kept in JA-10 centrifuge bottles. The cells are then pelleted by centrifuging them at 900 x g for 5 min at 4 °C in a Beckman JA-10 rotor.
- Chase
The cell pellet is resuspended in 5 ml of ice-cold HL-5 medium and then added to 100 ml of HL-5 medium kept in a 500 ml conical flask at 22 °C to initiate the chase. For chase, the cells are incubated at 22 °C and 150 rpm in a shaking incubator. To isolate early phagosomes, no chase is needed and one can proceed directly to step 10 whereas to obtain late phagosomes, the cells are chased for 45 min to complete the process of phagosome maturation. Before chase period ends, we generally take 50 µl of cells on a coverslip to check whether the cells have phagocytosed beads and to ensure that LBPs are motile in vivo (Video 2). Video 2. Motility of LBPs in Dictyostelium. 50 µl of cell suspension after the chase step was placed on a clean glass coverslip, allowed to adhere to the surface for 3-5 min and then imaged under 100x objective of a DIC microscope. The movie was acquired at 30 frames/sec using a custom program written on LabVIEW and later reduced to 10 frames/sec. Movie runs in real time. Latex beads (now LBPs), being highly refractile, can be easily distinguished from other endogenous organelles and show robust motion in vivo. - To stop the chase, the cell suspension is directly added to 330 ml of ice-cold Sorensen’s buffer kept in JA-10 centrifuge bottles. The cells are then pelleted by centrifuging them at 900 x g for 5 min at 4 °C in a Beckman JA-10 rotor.
- Removal of non-phagocytosed beads
After chase (or for early phagosomes after pulse), the cell pellet is washed thrice with ice-cold Sorensen’s buffer to remove any non-phagocytosed beads. During each washing step, the cell pellet is resuspended in 50 ml of Sorensen’s buffer and centrifuged at 900 x g for 5 min at 4 °C (see Note 11). - Cell lysis
After the final washing step, the cell pellet is weighed and then resuspended in a 1:1 (w/v) ratio of lysis buffer (LB/30% sucrose with inhibitors added and kept on ice). The cell suspension is then lysed by one passage through a preassembled syringe filter containing a 5 µm pore size membrane. The cell lysate is collected in a microfuge tube and kept on ice. Lysis efficiency is examined on a coverslip using a 20x objective of the cell culture microscope (Figure 2). For optimal yield, 70-80% lysis of cells is desirable (see Note 12).
Figure 2. Lysis of Dictyostelium cells using a 5 micron pre-assembled syringe filter. Cell suspension was spread on a clean coverslip before (A) and after (B) lysis and viewed under 20x objective of a negative phase contrast microscope. Intact cells appear dark (red arrowheads). Around 80-90% lysis is obtained using the preassembled filter. - High speed centrifugation
The cell lysate is then layered over a 1 ml cushion of LB/25% sucrose in an MLS-50 rotor tube and centrifuged in a Beckman MLS-50 rotor at 180,000 x g for 20 min at 4 °C (See Note 13). The LBPs, being more buoyant than other cell organelles and/or debris, do not pellet and are collected from the interface of the cell lysate and LB/25% sucrose as shown in Figures 3A and 3B. The top layer of the gradient is the cytosolic fraction. It is important to collect LBPs along with the cytosolic fraction otherwise their motility is compromised. - Collection of LBPs
A 1 ml syringe with needle is used to puncture the tube and phagosomes along with the cytosol are aspirated into the syringe (Figure 3B).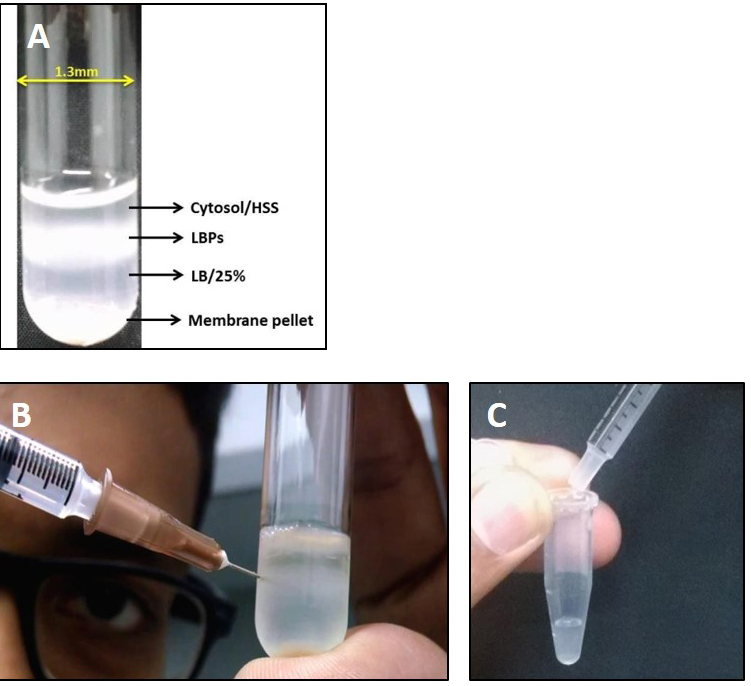
Figure 3. Extraction of LBPs from cell lysate. A. Image showing separation of LBPs on sucrose gradient post centrifugation. Membrane components are generally pelleted at the bottom whereas the cytosol is present at the top layer. LBPs float at the 15-25% sucrose interphase; B. Procedure for collection of LBPs from the gradient interphase using a syringe needle. It is important to remove the needle while transferring LBPs from the syringe into the collection tube as shown in C. - While transferring the aspirated solution from the syringe to a microfuge tube, it is important to remove the syringe needle (Figure 3C) otherwise the membrane might get disrupted.
- The phagosome/cytosol mixture is snap frozen in 36 µl aliquots in liquid nitrogen immediately after collection and stored in liquid nitrogen for up to a week. Each aliquot is rapidly thawed and used for one in vitro motility assay. Any unused thawed aliquot should be discarded.
Notes
- While ordering Dictyostelium cells, it is recommended to order them as spores plated on SM agar plate. Spores are more resistant to temperature fluctuations that might occur during transportation. The cells can easily be revived from the spores by picking and adding them to fresh HL-5 media kept at 22 °C.
- HL5 media is autoclaved at 120 °C, 20 psi pressure for 20 min. It is essential to remove the medium from the autoclave at the end of the 20 min. If left in the autoclave for prolonged duration, the media caramelizes (dark orange as opposed to a normal straw color) and is sub-optimal for cell culture usage.
- Following preparatory steps should be followed to perform the experiment in minimum amount of time:
- Keep centrifuges required at steps 7, 9, 10 and 12 at 4 °C.
- Dispense 100 ml HL5 media in a 500 ml conical flask (1 flask in case of early phagosome and 2 for late phagosomes) and pre-warm at 22 °C.
- Dispense 330 ml 1x Sorensen’s buffer in a JA-10 jar and keep on ice (1 in case of early phagosome and 2 for late phagosomes).
- Keep centrifuges required at steps 7, 9, 10 and 12 at 4 °C.
- Bead stock solution should not be more than a year old. Phagosomes prepared from older bead stocks show reduced or no motility. Also, bead solution to be used for phagocytosis is prepared fresh before extraction.
- Stock solution of pepstatin A (1 mg/ml) is prepared in methanol. It is highly recommended not to add glacial acetic acid to the solution as it compromises the motility of LBPs.
- It is important that the cell culture temperature is always maintained at 22 °C. Motility inside Dictyostelium cells is highly sensitive to temperature and a change of even 2-3 °C from the optimum can disrupt motility.
- It is important to maintain the range of optimal cell density for the extraction protocol. Over-confluent cells show reduced motility whereas cell count below the lower limit often leads to difficulty during syringe lysis of cells. In case of lower cell density (3-4 x 106 cells/ml), two 100 ml suspension cultures can be used provided cells from both the cultures show robust intracellular motility. Using cells from an over-confluent suspension (density < 8 x 106 cells/ml) is not recommended since the cells will begin the developmental phase of their life cycle. In vitro motion has only been characterized for LBPs isolated from vegetatively growing cells.
- Bead washing with HL-5 medium serves a dual purpose: (a) It removes azide and other preservatives which are present in the bead stock solution. (b) It coats the beads with a layer of HL-5 medium facilitating the process of bead uptake by cells during the phagocytosis step. Skipping this washing step therefore would lead to suboptimal phagocytosis and compromise the yield. Also, azide in the stock solution could potentially kill phagosome motility.
- Synchronization step at 4 °C causes attachment of beads onto the cell surface but does not allow the initiation of phagocytosis. Thus, when phagocytosis is allowed to begin at 22 °C, it ensures a synchronized uptake of beads by cells. This is critical to isolate phagosomes in a stage-specific manner.
- It is crucial to strictly adhere to the pulse time duration, especially while isolating early phagosomes. In Dictyostelium, the early stage of phagosome maturation lasts for 5 min and any delay beyond this time point will cause a switch to late phagosome stage, which is also reflected in the phagosome motility.
- Improper washing of cells after pulse/chase leads to the presence of non-phagocytosed beads in the final phagosome extract. These beads cannot be distinguished from phagosomes under DIC microscope and are immotile during the motility assay. Hence, their presence can lead to a false conclusion of compromised phagosome motility.
- Cell lysis is the most crucial step in the extraction protocol. The success of this step is directly correlated with phagosome yield and the quality of phagosome motility. Therefore, it is imperative that this step be performed carefully.
- The total volume of the cushion and the cell lysate should at least be half of the maximum capacity of the ultra-centrifugation tube (for MLS-50, the capacity is 5 ml). Under circumstances when the above condition is not met, the cushion volume can be increased to 2 ml.
Recipes
- Sorensen’s buffer
15 mM KH2PO4
2 mM Na2HPO4
Adjust pH to 6.0
Note: This buffer is used for washing of cells to remove media and non-phagocytosed beads. We prepare a 10x stock of this buffer, which is autoclaved and stored at 4 °C. Before each extraction, 1 L of 1x buffer is prepared and chilled for an hour before usage. - Cell lysis buffer (LB/30% sucrose)
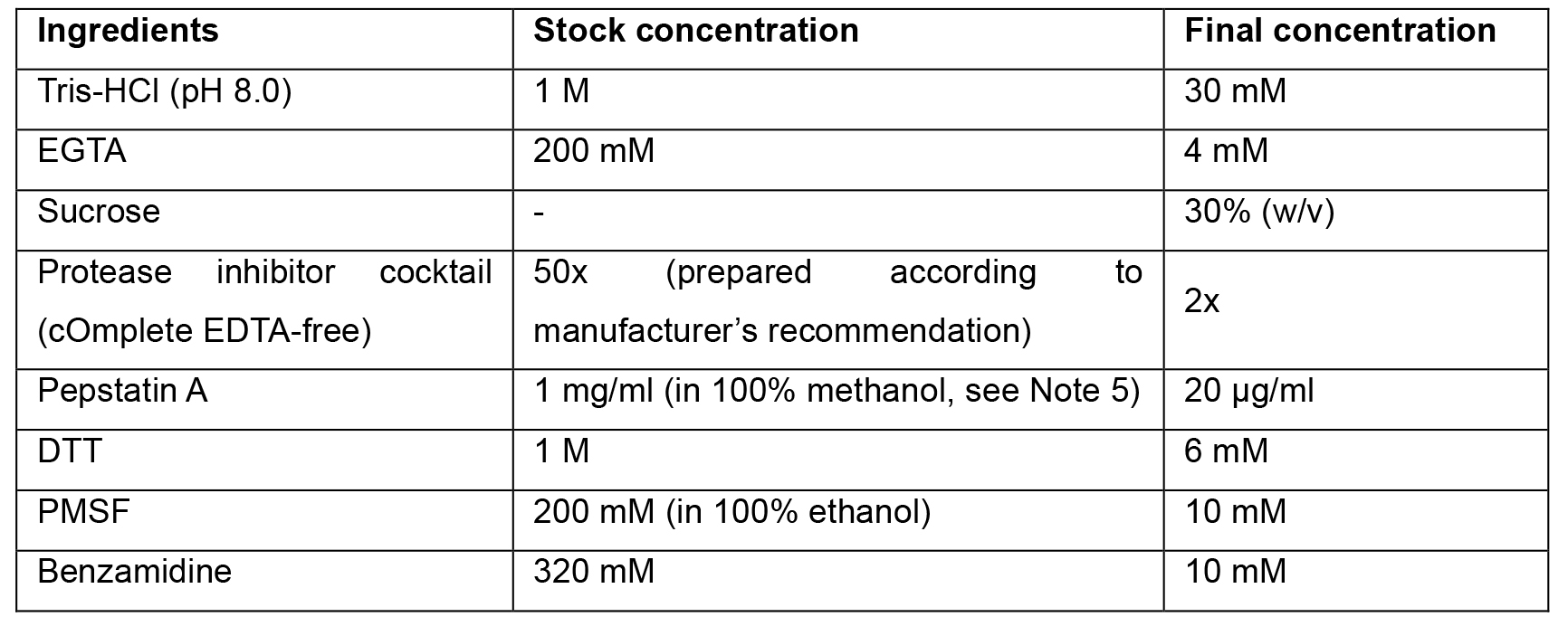
Note: All stock solutions are prepared in ddH2O water if not stated otherwise. Stock solutions of protease inhibitor cocktail, pepstatin A and DTT stored at -20 °C; PMSF and Benzamidine stocks are prepared fresh. All of these are added to the buffer prior to the cell lysis step. It is important to carefully monitor the pH of this buffer. Deviations from pH 8.0 are detrimental for phagosome motility. - Centrifugation cushion buffer (LB/25% sucrose)
Same composition as the cell lysis buffer except that it contains 25% (w/v) sucrose instead of 30% (w/v) sucrose
Acknowledgments
We acknowledge funding through an International Senior Research Fellowship from the Wellcome Trust UK (grant WT079214MA). We also acknowledge funding from the Wellcome Trust – Department of Biotechnology Alliance, India (Senior Fellowship grant IA/S/11/2500255 to RM and Early career Fellowship grant IA/E/15/1/502298 to PS).
References
- Barak, P., Rai, A., Dubey, A. K., Rai, P. and Mallik, R. (2014). Reconstitution of microtubule-dependent organelle transport. Methods Enzymol 540: 231-248.
- Blocker, A., Severin, F. F., Burkhardt, J. K., Bingham, J. B., Yu, H., Olivo, J. C., Schroer, T. A., Hyman, A. A. and Griffiths, G. (1997). Molecular requirements for bi-directional movement of phagosomes along microtubules. J Cell Biol 137(1): 113-129.
- Gotthardt, D., Dieckmann, R., Blancheteau, V., Kistler, C., Reichardt, F. and Soldati, T. (2006). Preparation of intact, highly purified phagosomes from Dictyostelium. Methods Mol Biol 346: 439-448.
- Harrison, R. E., Brumell, J. H., Khandani, A., Bucci, C., Scott, C. C., Jiang, X., Finlay, B. B. and Grinstein, S. (2004). Salmonella impairs RILP recruitment to Rab7 during maturation of invasion vacuoles. Mol Biol Cell 15(7): 3146-3154.
- Harrison, R. E. and Grinstein, S. (2002). Phagocytosis and the microtubule cytoskeleton. Biochem Cell Biol 80(5): 509-515.
- Rai, A., Pathak, D., Thakur, S., Singh, S., Dubey, A. K. and Mallik, R. (2016). Dynein clusters into lipid microdomains on phagosomes to drive rapid transport toward lysosomes. Cell 164: 722-734.
- Sun, J., Deghmane, A. E., Soualhine, H., Hong, T., Bucci, C., Solodkin, A. and Hmama, Z. (2007). Mycobacterium bovis BCG disrupts the interaction of Rab7 with RILP contributing to inhibition of phagosome maturation. J Leukoc Biol 82(6): 1437-1445.
- Vieira, O. V., Botelho, R. J. and Grinstein, S. (2002). Phagosome maturation: aging gracefully. Biochem J 366(Pt 3): 689-704.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
D’Souza, A., Sanghavi, P., Rai, A., Pathak, D. and Mallik, R. (2016). Isolation of Latex Bead Phagosomes from Dictyostelium for in vitro Functional Assays. Bio-protocol 6(23): e2056. DOI: 10.21769/BioProtoc.2056.
Category
Cell Biology > Organelle isolation > Phagosome
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.