- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

In vitro Autophosphorylation and Phosphotransfer Assay of Cyanobacterial Histidine Kinase 2

Published: Vol 6, Iss 23, Dec 5, 2016 DOI: 10.21769/BioProtoc.2036 Views: 11679
Reviewed by: Maria SinetovaAnna A. ZorinaTatsuki Kunoh
Original research article
The authors used this protocol in:

Feb 2016
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
This is a detailed protocol of an autophosphorylation and phosphotransfer activities of Synechocystis sp. PCC 6803 full-length Histidine Kinase 2 (Hik2) protein described by Ibrahim et al., 2016. In this protocol, radioactively labelled ATP was used to study an autophosphorylation and phosphotransfer activity of the full-length Hik2 protein.
Keywords: Histidine Kinase 2Background
Protein phosphorylation is an important post-translational modification of proteins that takes place in every living organism. The activity of protein kinases, the enzyme that catalyses the phosphorylation of proteins, was first described by Burnett and Kennedy in 1954, where they showed phosphorylation of casein by a liver enzyme (Burnett and Kennedy, 1954). However, its significance was not appreciated until the 1970s and 1980s (Cohen, 2002). The transfer of the γ-phosphate from an ATP molecule to proteins can be studied using coupled assays or directly with radioactively labelled ATP. Kinase assays based on incorporation of 32P can easily be followed by autoradiography, whereas coupled assays require monitoring of indirect reporter enzyme-catalysed colorimetric or chemiluminescence signals. The work presented here was conducted using radioactive ATP. Serine/threonine-type protein kinases dominate in eukaryotes, while in prokaryotes histidine kinases are the primary protein kinases involved in signal transduction. A histidine kinase catalyses the transfer of only γ-phosphate from an ATP molecule to its conserved histidine residue and transfers phosphoryl group to its response regulator (see Figure 1), but cannot catalyse the transfer of α-phosphate of ATP (Pernestig et al., 2001). Therefore [α-32P]ATP can be used as a negative control when characterising the autophosphorylation activity of putative histidine kinases.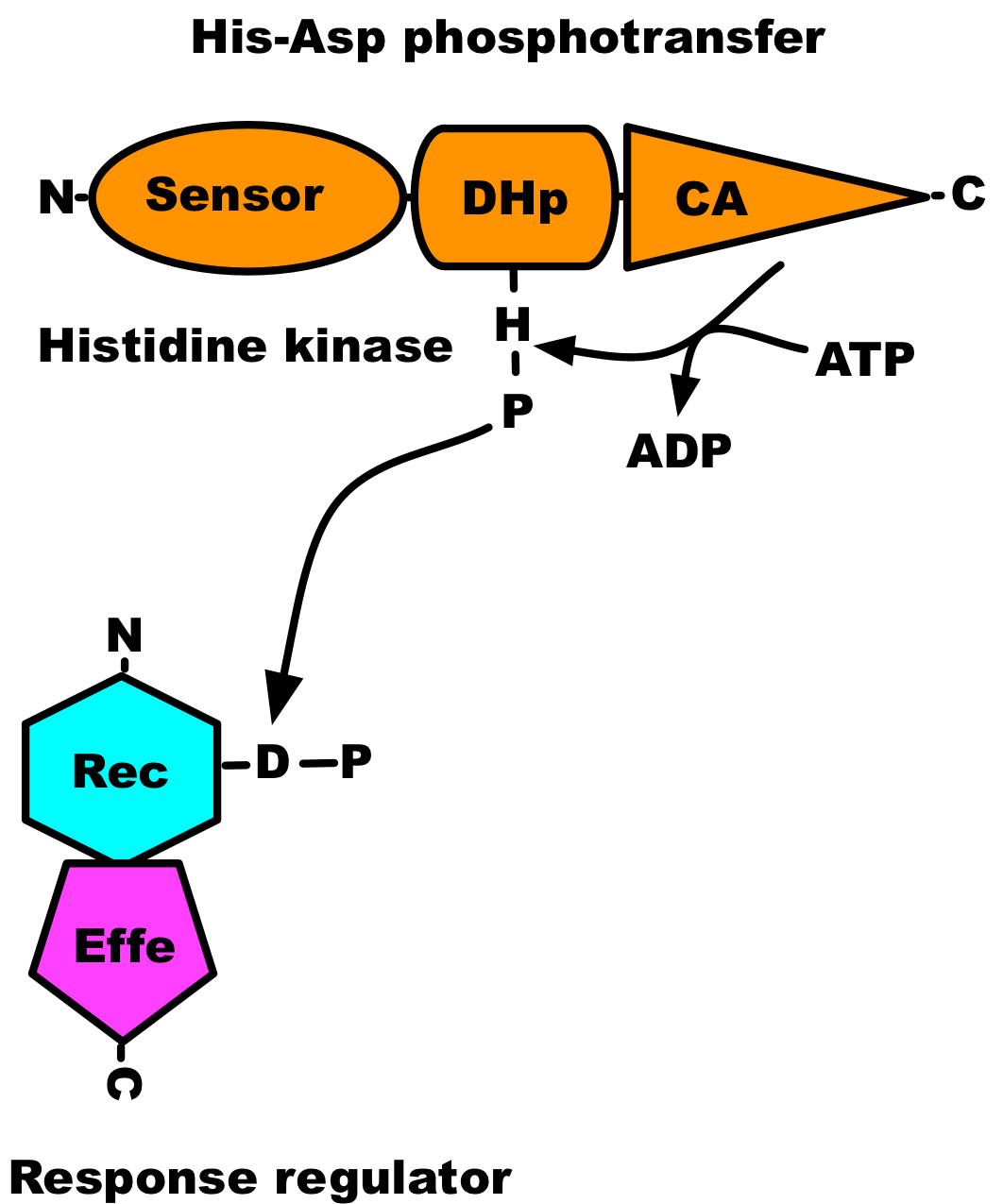
Figure 1. Domain architecture of two-component system. The sensor domain is indicated by oval, the dimerisation and phosphoaccepting (DHp) domain by a cylinder, and the catalytic and ATP-binding (CA) domain by a triangle; receiver (Rec) domain by a hexagon; effector (Effe) domain by a pentagon.
Materials and Reagents
- Eppendorf tubes
- 50 ml Falcon tubes
- Pipette tips
- Chelating Sepharose Fast Flow (GE Healthcare, catalog number: 17057501 )
- PD-10 desalting columns (GE Healthcare, catalog number: 17085101 )
- 1 ml cuvette
- Polyethylene bags (Thermo Fisher Scientific, Fisher Scientific, catalog number: 01817200 )
- BL21-DE3 E.coli cells containing Hik2, Rre1, and RppA clones. Each protein should be prepared fresh for each assay.
- pET-21b vector (Invitrogen)
- One Shot® TOP10 chemically competent E. coli (Thermo Fisher Scientific, InvitrogenTM, catalog number: C404006 )
- BL21-(DE3) chemical competent cells
- NdeI endonuclease (New England BioLabs, catalog number: R0111S )
- XhoI endonuclease (New England BioLabs, catalog number: R0146S )
- KpnI
- Primers were purchased from Eurofins MWG Operon, Germany.
- Deoxynucleoside triphosphate set (Sigma-Aldrich, catalog number: DNTP-RO )
- Phusion® high-fidelity DNA polymerase (New England BioLabs, catalog number: M0530S )
- RNase/DNase free water
- GeneJET Gel Extraction Kit (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: K0691 )
- Tris-HCl
- Bovine serum albumin (BSA) (New England BioLabs, catalog number: B9000S )
- DNA loading dye (6x) (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: R0611 )
- Agarose
- Fermentas Gel Extraction Kit
- T4-ligase (New England BioLabs, catalog number: M0202S )
- Ampicillin sodium salt (Sigma-Aldrich, catalog number: A9518-25G )
- Isopropyl β-D-1-thiogalactopyranoside (IPTG) (Melford Laboratories, catalog number: MB1008 )
- Imidazole
- Bradford reagent (Sigma-Aldrich, catalog number: B6916-500ML )
- 500 μCi [γ-32P]-ATP (6,000 Ci mmol-1) (PerkinElmer, catalog number: NEG502Z500UC )
- Adenosine 5’-triphosphate disodium salt hydrate (Sigma-Aldrich, catalog number: A2383-5G )
- Luria broth (LB), low salt, granulated (Melford Laboratories, catalog number: GL1703 )
- KCl
- MgSO4
- MgCl2
- Glucose
- NaCl
- PMSF
- Glycerol
- SDS
- β-2-mercaptoethanol
- 30% acrylamide/bis-acrylamide
- APS
- TEMED
- Precision Plus Protein All Blue Standards (Bio-Rad Laboratories, catalog number: 161-0373 )
- LB medium (see Recipes)
- Super optimal broth with catabolic repressor (SOC) (see Recipes)
- Lysis buffer/wash buffer 1 (see Recipes)
- Wash buffer 2 (see Recipes)
- Wash buffer 3 (see Recipes)
- Elution buffer (see Recipes)
- PD-10 desalting column equilibration buffer (see Recipes)
- 5x kinase reaction buffer (see Recipes)
- 5x ATP mix (see Recipes)
- SDS-PAGE Laemmli sample buffer (see Recipes)
- SDS-PAGE (see Recipes)
- 1x SDS-PAGE running buffer (see Recipes)
Equipment
- PCR machine
- Pipette shield
- 2 L Erlenmeyer flask
- Bottle assembly, J-Lite PC-1000, polycarbonate (Beckman Coulter, catalog number: 363676 )
- Backment Coulter AvantiTM J-30I centrifuge (Beckman Coulter, model: Avanti J-30I )
- EmulsiFlex-C3 homogenizer (Abestin, model: EmulsiFlex-C3 )
- Bottle assembly, polycarbonate, 50 ml (Beckman Coulter, order number: 357000 )
- Heating block
- Phosphorimager (Molecular Dynamics)
- Fume-hood
- GM counters
- Perspex Eppendorf holders
- JA-30.5 Ti rotor (Beckman Coulter, model: JA-30.5 Ti Rotor )
- JLA-9.1000 rotor, fixed angle (Beckman Coulter, catalog number: 366754 )
- Gilson pipettes
- Plexiglas shielding
- Mini-PROTEAN® Electrophoresis system (Bio-Rad Laboratories, catalog number: 1658000EDU )
- Bio-Rad PowerPac (Bio-Rad Laboratories, catalog number: 1645050 )
- Phosphor cassette (Molecular Dynamics)
- Phosphor plate (Molecular Dynamics)
- Image Eraser (Molecular Dynamics)
Software
- ImageQuant software (Molecular Dynamics)
Procedure
- Cloning of Hik2, Rre1 and RppA genes
- Polymerase Chain Reaction (PCR): Perform PCR reaction for coding sequences corresponding to the full-length Synechocystis sp. PCC6803 Hik2 (slr1147), Rre1 (slr1783), and RppA (sll0797) from Synechocystis sp. PCC 6803 genomic DNA using primer pairs listed in Table 1. Digest the PCR product of full-length Hik2 (Hik2) with NdeI and XhoI (New England BioLabs) and clone into a pET-21b vector (Invitrogen). Digest Rre1 and RppA with KpnI and XhoI endonucleases (New England BioLabs) and clone into pETG-41A (EMBL) expression vector. Prepare the following PCR reaction in a total volume of 50 µl:
- Add 1 µl of dNTPs (deoxynucleoside triphosphates, 10 mM each), final concentration is 200 µM each.
- 4 µl of the 10x HF Phusion DNA polymerase reaction buffer.
- 2.5 µl of forward primer (final concentration of 0.5 µM).
- 2.5 µl of reversed primer (final concentration of 0.5 µM).
- 5 ng of template DNA.
- 0.5 µl of Phusion DNA polymerase (final concentration of 0.01 units).
- Adjust the volume to 50 µl with RNase/DNase free water.
- Incubate in the PCR machine. For PCR programme, see Table 2.
- Purify the PCR amplicon from enzymatic reaction buffers using GeneJET Gel Extraction Kit and elute the DNA sample in 50 µl of Tris-HCl (pH 8.0).
Table 1. Primer pairs used for cloning Hik2, Rre1, and RppA genes
Note: Base pairs in lower case are restriction site overhangs.
Table 2. PCR cycling programme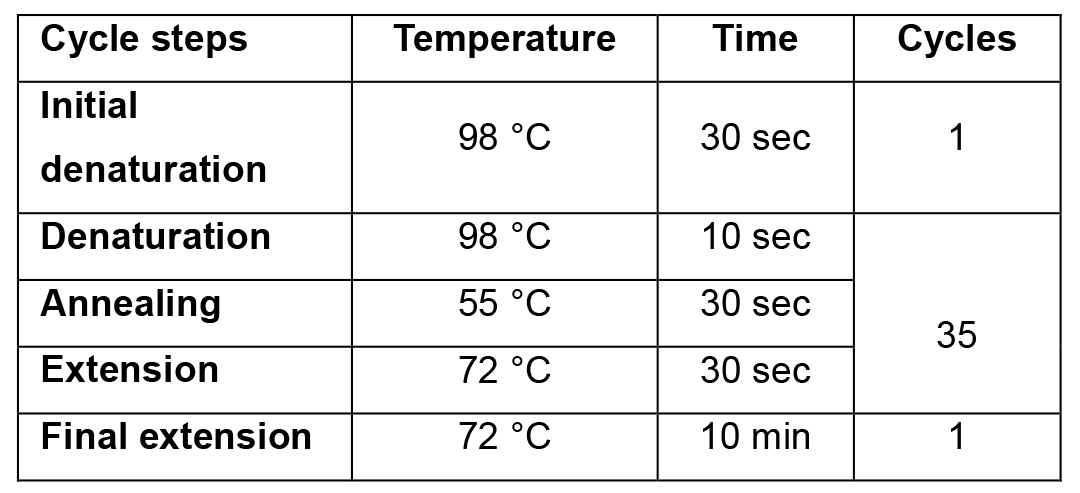
- Add 1 µl of dNTPs (deoxynucleoside triphosphates, 10 mM each), final concentration is 200 µM each.
- Restriction endonuclease digest: Carry out the following double digestion reaction in a total volume of 50 µl:
- To a 1.5 ml Eppendorf tube, add 8 µg of PCR product or 4 µg of plasmid.
- 1 µl of the appropriate endonucleases (40 units final).
- 0.25 µl of the 20 mg ml-1 BSA (final concentration of 100 µg ml-1).
- 5 µl of 10-fold concentrated NEB-buffer.
- Adjust the volume to 50 µl with RNase/DNase free water.
- Incubate reactions at 37 °C for 3 h.
- Add 10 µl of 6 fold concentrated DNA loading dye and loaded onto an agarose 1% gel.
- Once the DNA fragments are separated on agarose gel, cut the bands and purified from the gel using Fermentas Gel Extraction Kit.
- To a 1.5 ml Eppendorf tube, add 8 µg of PCR product or 4 µg of plasmid.
- Ligation: Carry out the following ligation reaction in a total reaction volume of 20 µl.
- Mix the double digested plasmid and PCR product in 1:3 molar ratios, respectively.
- Add 1 µl of T4-ligase (20 units final).
- 2 µl of 10x T4 ligase buffer.
- Adjust the volume to 20 µl with DNase/RNase-free water.
- Incubate reaction at 25 °C for 15 min or at 16 °C overnight.
- Mix the double digested plasmid and PCR product in 1:3 molar ratios, respectively.
- Recombinant transformation
- To a pre-chilled 1.5 ml Eppendorf tube, aliquot 50 μl One Shot TOP10 chemical competent cells.
- Add 0.2 μl (~2 ng) of ligation product into the cells.
- Incubated on ice for 30 min.
- Heat shock at 42 °C for 90 sec and incubated on ice for further 60 sec.
- Diluted to 1:10 by adding 950 μl of preheated (at 37 °C) SOC medium.
- Incubated at 37 °C whilst agitating at 200 rpm for 1 h.
- Briefly spin and discard ~800 µl of the medium. Resuspend the pellet with the remaining medium and plate onto agarose LB-Amp plate.
- Incubated at 37 °C overnight.
- The following day, inoculate a single colony into 5 ml LB-medium containing 100 μl ml-1 ampicillin and incubated at 37 °C overnight whilst agitating at 200 rpm.
- Extract the clones and confirm the presence of clone by double digesting with the above restriction and sequencing.
- To a pre-chilled 1.5 ml Eppendorf tube, aliquot 50 μl One Shot TOP10 chemical competent cells.
- Transform the above clone into BL21-(DE3) chemical competent cells as above for recombinant protein production.
- Polymerase Chain Reaction (PCR): Perform PCR reaction for coding sequences corresponding to the full-length Synechocystis sp. PCC6803 Hik2 (slr1147), Rre1 (slr1783), and RppA (sll0797) from Synechocystis sp. PCC 6803 genomic DNA using primer pairs listed in Table 1. Digest the PCR product of full-length Hik2 (Hik2) with NdeI and XhoI (New England BioLabs) and clone into a pET-21b vector (Invitrogen). Digest Rre1 and RppA with KpnI and XhoI endonucleases (New England BioLabs) and clone into pETG-41A (EMBL) expression vector. Prepare the following PCR reaction in a total volume of 50 µl:
- Protein production and purification
- Inoculate a single colony of BL21-DE3 E. coli cells containing Hik2 clone into 10 ml Luria broth (LB) growth medium (Sambrook et al., 1989) supplemented with 100 μg ml-1 ampicillin. Incubate at 37 °C while shaking at 200 rpm.
- The following morning, in 2 L Erlenmeyer flask, dilute the above starter culture at 1:100 in 1 L LB medium supplemented with 100 μg ml-1 ampicillin.
- Incubate at 37 °C while shaking at 200 rpm. Monitor the optical density at 600 nm until it reaches ~0.55.
- Induce protein expression with 0.5 mM IPTG.
- Incubate the induced bacterial culture for a further 16 h at 16 °C while shaking at 150 rpm.
- Transfer the bacterial cells into 1 L polycarbonate bottle and harvest by centrifugation at 9,000 x g for 10 min at 4 °C.
- Resuspend the pellet in 20 ml of the lysis buffer/wash buffer 1 per 1 L of culture.
- Lyse the cells by passing through EmulsiFlex-C3 homogenizer two times and transfer the lysate to 50 ml polycarbonate bottle.
- Separate the lysate by centrifugation at 39,000 x g for 20 min at 4 °C. And transfer the supernatant to 50 ml Falcon tube.
- Equilibrate the Ni2+ affinity chromatography column with 5 ml (5-bead volume) of the lysis buffer.
- Apply the supernatant from step B9 to the equilibrated Ni2+ affinity chromatography column.
- Wash the column with 30 ml of lysis buffer/wash buffer 1 containing 25 mM imidazole.
- Wash with 30 ml of wash buffer 2 containing 50 mM imidazole.
- Wash with 5 ml of wash buff 3 containing 100 mM imidazole.
- Elute the protein with 1 ml of elution buffer. Repeat this step B15 three more times.
- Inoculate a single colony of BL21-DE3 E. coli cells containing Hik2 clone into 10 ml Luria broth (LB) growth medium (Sambrook et al., 1989) supplemented with 100 μg ml-1 ampicillin. Incubate at 37 °C while shaking at 200 rpm.
- Desalting: prepare the following steps in the cold room
- Remove the top cap of PD-10 desalting column and pour off the column storage solution.
- Cut the sealed end of the column at notch.
- Fill up the column with PD-10 desalting/equilibration buffer and allow the equilibration buffer to enter the packed bed completely.
- Repeat step C3 4 times.
- Discard the flow-through.
- Add maximum 2.5 ml of sample to the column. For sample volumes less than 2.5 ml, add desalting/equilibration buffer to adjust the volume to 2.5 ml after the sample has entered the packed bed completely.
- Let the sample or desalting/equilibration buffer enter the packed bed completely.
- Discard the flow-through.
- Place a 1.5 ml Eppendorf tube for sample collection under the column.
- Elute with 3.5 ml desalting/equilibration buffer and collect 3 times 1 ml and finally 0.5 ml of eluate.
- Use the eluate for autophosphorylation immediately.
- Remove the top cap of PD-10 desalting column and pour off the column storage solution.
- Determination of protein concentration using Bradford assay
- Add 33 μl of Hik2 protein solution into 1.5 ml Eppendorf tube. To the second 1.5 ml Eppendorf tube add 33 µl of elution buffer and label this tube as blank.
- To each tube, add 1 ml of Bradford reagent and incubated at room temperature (~22 °C) for 10 min.
- Transfer the samples into 1 ml cuvette.
- Measure the absorbance for each sample at 595 nm and calculate protein concentration from BSA standard curve.
- Add 33 μl of Hik2 protein solution into 1.5 ml Eppendorf tube. To the second 1.5 ml Eppendorf tube add 33 µl of elution buffer and label this tube as blank.
- In vitro autophosphorylation assay: prepare the following in ice
- Add 2 μM of purified Hik2 protein to 1.5 ml Eppendorf tube.
- Add 5 μl of 5-fold concentrated kinase reaction buffer.
- Adjust the volume to 20 μl with dH2O.
- Equilibrate the samples to 22 °C in heating block.
- Mix gently by vortexing for 1-2 sec.
- Initiate the autophosphorylation reaction by the addition of 5 μl of 5-fold concentrated ATP mix. Immediately start the stopwatch and let the autophosphorylation reaction proceed for 15 sec at 22 °C.
- Terminate the reaction by adding 6 μl of 5-fold concentrated Laemmli sample buffer (Laemmli, 1970).
- Separate the reaction products on a 12% reducing SDS-PAGE.
- Once protein separation is complete, rinse the gel 2-3 times with SDS running buffer and transfer the gel into a sealed polyethylene bag.
- Place the sealed bag against the phosphor screen and expose overnight.
- Visualise the incorporated γ-phosphate is using autoradiography.
- Add 2 μM of purified Hik2 protein to 1.5 ml Eppendorf tube.
- Phosphotransfer analysis
- Carry out an autophosphorylation reaction by mixing 12 µM of Hik2 protein in a total reaction volume of 150 µl containing 30 µl of the 5x concentrated kinase reaction buffer and 30 µl of 5x concentrated ATP mix.
- Incubate the reaction at 30 °C for 10 min.
- In the meantime, prepare 25 µM of each of the response regulator (MBP-Rre1 or MBP-RppA) in a total volume of 62.5 µl containing 12.5 µl of the 5x concentrated kinase reaction buffer.
- Prepare a control reaction lacking response regulator in the same way, except that replace response regulator protein solution with an equal volume of water.
- Remove 12.5 µl of the autophosphorylated Hik2 protein and add 3 µl of Laemmli sample buffer to stop the reactions. Label this tube as T0.
- For each phosphotransfer reaction, take 62.5 µl of the autophosphorylated radiolabeled Hik2 protein from step F1 and mix it with 62.5 µl of the response regulator or with the water control from steps F3 and F4. Kinase and response regulator should be present at a concentration of 1 µM and 5 µM, respectively.
- Mix briefly by vortexing and incubated at 30 °C.
- Remove 25 µl samples at 20, 40, 60, and 90 min and add 6 µl of Laemmli sample buffer to stop the reactions.
- Resolve the proteins on 15% SDS-PAGE and visualized the presence of γ-32P using autoradiography.
Table 3. Protein molecular weight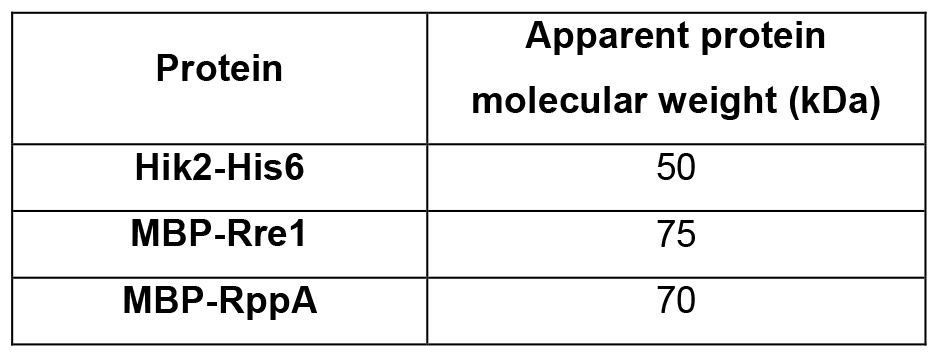
Data analysis
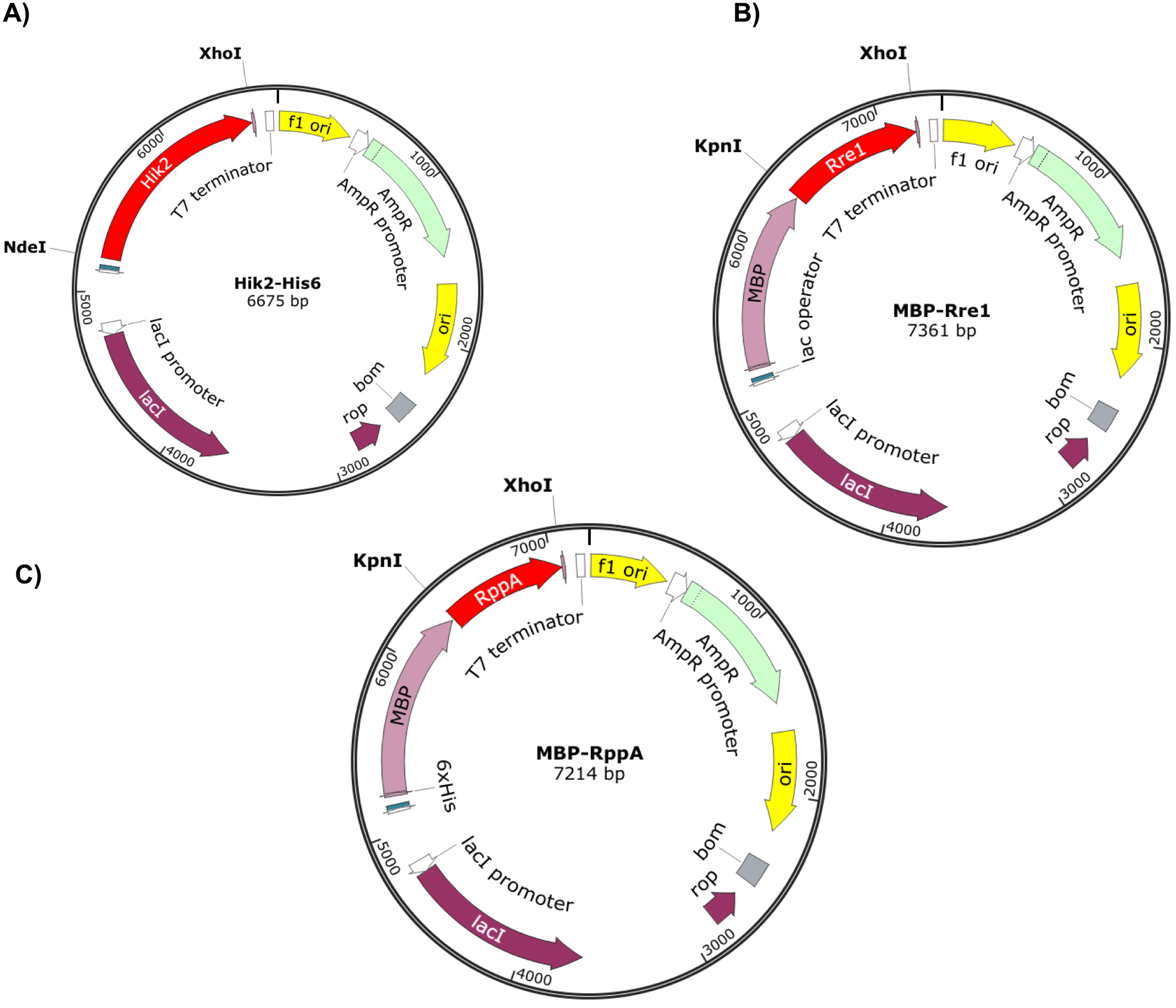
Figure 2. Recombinant vectors containing Hik2, Rre1 and RppA genes. The full-length Hik2 (A), Rre1 (B), and RppA (C) open reading frames are shown by red arrow. The C-terminal poly-histidine (His-tag) and the N-terminal MBP tags are shown by magenta. The f1 and Ori origins of replication are shown by yellow colour; an ampicillin resistant gene is shown by light green; purple and dark green shows the lacI and T7/lac promoters, respectively. The direction of arrow indicates the direction of translation of genes. Vector maps were drawn using SnapGene.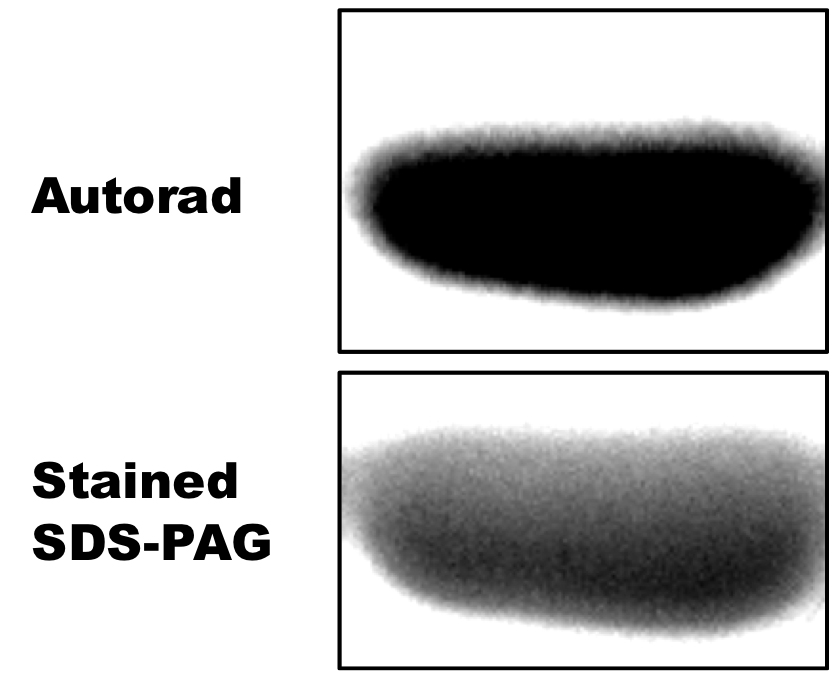
Figure 3. Autophosphorylation activity of Hik2. An autoradiograph (autorad) showing autophosphorylated recombinant Hik2 protein. 2 μM of purified Hik2 protein was assayed in the presence of 2.5 μCi ATP at 22 °C. Hik2 protein was then resolved on a 12% SDS-PAGE and the incorporation of 32P was monitored by autoradiography.
Figure 4. Phosphotransfer activity of Hik2 to Rre1 and RppA response regulators. An autoradiograph (autorad) showing autophosphorylated recombinant Hik2 protein. 2 μM of purified Hik2 protein was assayed in the presence of 2.5 μCi ATP at 22 °C. Hik2 protein was then resolved on a 12% SDS-PAGE and the incorporation of 32P was monitored by autoradiography.
Notes
- Radiation Hazards: possible routes of internal and external contamination
- Internal contamination could occur while mixing samples by vortices. Samples should be mixed in the fume-hood to reduce the potential hazard posed by volatile vapour.
- Aerosol: The isotope stock should be opened inside a fume-hood to prevent inhalation of aerosol.
- Spillage: Spillage should be monitored using GM counters and the use of the laboratory spill kit.
- External contamination may also occur while handling the radioactive samples. Appropriate shielding materials should be used to reduce external contamination. All radiation samples should be used behind Perspex shielding.
- In addition, solid type Perspex Eppendorf holders should be used to further reduce exposure.
Recipes
- LB medium
Dissolve 20 g of LB broth, low salt (Lennox L Broth), granulated in 1 L of dH2O - Super optimal broth with catabolic repressor (SOC)
To 10 ml LB-broth filter sterilised:
2.5 mM KCl
10 mM MgSO4
10 mM MgCl2
20 mM glucose - Lysis buffer/wash buffer 1
300 mM NaCl
20 mM Tris-HCl, pH 7.4
25 mM imidazole
1 mM PMSF - Wash buffer 2
300 mM NaCl
20 mM Tris-HCl, pH 7.4
50 mM imidazole - Wash buffer 3
300 mM NaCl
20 mM Tris-HCl, pH 7.4
100 mM imidazole - Elution buffer
300 mM NaCl
20 mM Tris-HCl, pH 7.4
500 mM imidazole - PD-10 desalting column equilibration buffer
Tris-HCl (10 mM final, pH 7.4) - 5x kinase reaction buffer (1 ml)
250 mM Tris-HCl (pH 7.5)
250 mM KCl
50% glycerol
50 mM MgCl2 - 5x ATP mix
2.5 mM ATP
25 μCi [γ-32P]-ATP (3,000 Ci mmol-1) - 5x SDS-PAGE Laemmli sample buffer
164.5 mM Tris-HCl, pH 6.8
26.3 % (w/v) glycerol
5.25 % (w/v) SDS
25 % (v/v) β-2-mercaptoethanol
0.025% bromophenol blue - SDS-PAGE
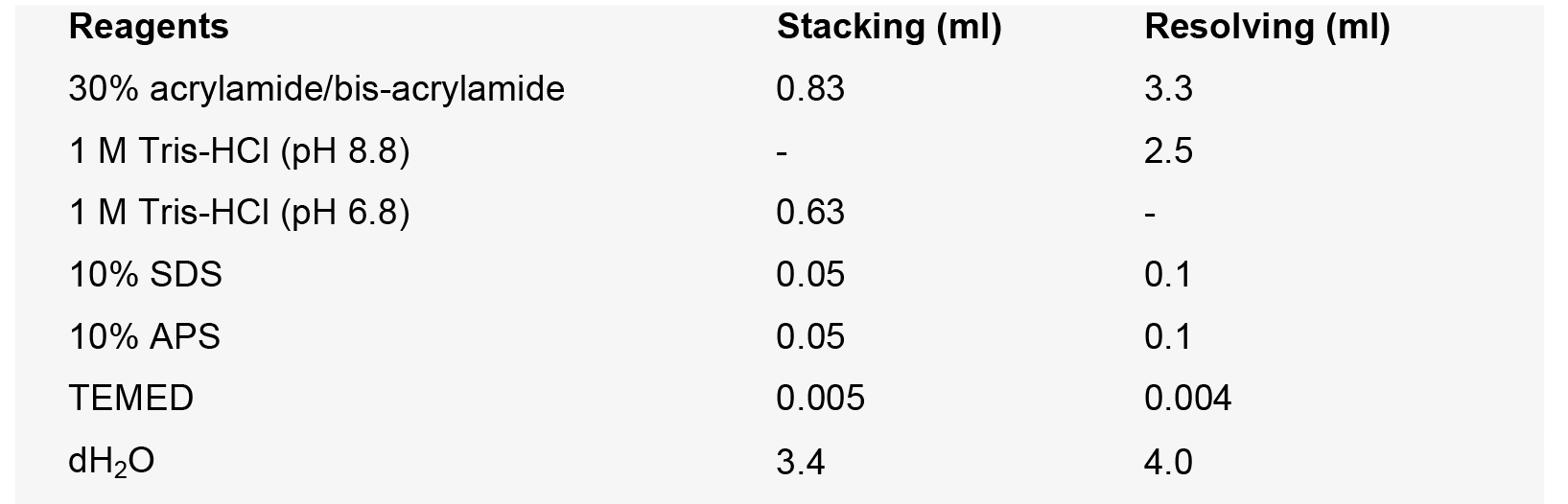
- 1x SDS running buffer (1 L)
0.025 M Tris base
0.192 M glycine
0.1% SDS
The pH as it is should be 8.3. No need to adjust
Acknowledgments
This protocol was adapted from an earlier report (Ibrahim et al., 2016). I.M.I thanks Queen Mary University of London for a graduate teaching studentship. The author would like to express his sincere gratitude to Prof. John F. Allen and Dr. Sujith Puthiyaveetil for their comments.
Conflict of Interest Statement: The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Burnett, G. and Kennedy, E. P. (1954). The enzymatic phosphorylation of proteins. J Biol Chem 211(2): 969-980.
- Cohen, P. (2002). The origins of protein phosphorylation. Nat Cell Biol 4(5): E127-130.
- Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259): 680-685.
- Ibrahim, I. M., Puthiyaveetil, S. and Allen, J. F. (2016). A two-component regulatory system in transcriptional control of photosystem stoichiometry: Redox-dependent and sodium ion-dependent phosphoryl transfer from cyanobacterial histidine kinase Hik2 to response regulators Rre1 and RppA. Front Plant Sci 7: 137.
- Pernestig, A. K., Melefors, O. and Georgellis, D. (2001). Identification of UvrY as the cognate response regulator for the BarA sensor kinase in Escherichia coli. J Biol Chem 276(1): 225-231.
- Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989). Molecular cloning: A laboratory manual. Second edition. Cold Spring Harbor, New York: Greene Publishing Associates and John Wiley & Sons. pp: 1126.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Ibrahim, I. M. (2016). In vitro Autophosphorylation and Phosphotransfer Assay of Cyanobacterial Histidine Kinase 2. Bio-protocol 6(23): e2036. DOI: 10.21769/BioProtoc.2036.
Category
Microbiology > Microbial signaling > Phosphorylation
Molecular Biology > Protein > Phosphorylation
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.