- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Displacement-based ELISA: Quantifying Competition between Two Binding Partners for Interaction with a His-tagged Ligand Immobilized on a Ni2+-NTA Plate
Published: Vol 6, Iss 5, Mar 5, 2016 DOI: 10.21769/BioProtoc.1745 Views: 10109
Reviewed by: Valentine V TrotterAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Related protocols
Abstract
The displacement assay was designed to quantify the direct competition between two homologous ribosomal proteins from Mycobacterium tuberculosis, S18-1 and S18-2, for interaction with their cognate binding partner, ribosomal protein S6 (Prisic et al., 2015). The S18 proteins were dialyzed in two physiologically relevant conditions (i.e. in the presence of Zn2+ or with EDTA to chelate Zn2+) and then allowed to compete for binding to S6 which was maintained in limiting concentration. The result was obtained through an ELISA, where S6-His is first bound to a Ni2+-NTA plate, followed by addition of S18-2 in excess to S6, then by addition of increasing concentrations of S18-1. The percentage of S18-2 that remained bound to S6 was quantified with antibodies specific to the S18-2 protein and secondary antibodies, in chemiluminescent ELISA. In this way displacement of S18-2 protein by the S18-1 protein was reported as a percentage of the full strength signal achieved through saturation of S6 with S18-2. At its foundation, this method exploits a native protein-protein interaction and could be applied to other systems where two or more proteins compete for binding to a target ligand as above.
Keywords: ELISAMaterials and Reagents
- HisPurTM Cobalt Spin Columns (Thermo Fisher Scientific, catalog number: 89969 )
- Pierce™ Polyacrylamide Spin Desalting Columns, 7 K MWCO, 0.7 ml (Thermo Fisher Scientific, catalog number: 89849 )
- 5 PRIMETM Ni2+-NTA HisPrimeTM plates (Thermo Fisher Scientific, catalog number: 2400730 )
- ProTEV Plus (Promega Corporation, catalog number: V6101 )
- cOmplete His-Tag Purification Resin (Sigma-Aldrich, Roche Diagnostics, catalog number: 05893682001 )
- BioRad Protein Concentration Assay (Bio-Rad Laboratories, catalog number: 5000002 )
- Albumin, Bovine, Fraction V (pH 7) (BSA) (Affymetrix, catalog number: 10857 50 gm )
- Rabbit Polyclonal Antibody raised to peptide antigen from competing protein (i.e. S18-2) (NeoPeptide, custom made against peptide PGQDRQRRAALCP)
- Goat Anti-Rabbit IgG, Peroxidase Conjugated Secondary Antibody (Thermo Fisher Scientific, PierceTM, catalog number: 31460 )
- SuperSignalTM West Pico Chemiluminescent Substrate (Thermo Fisher Scientific, catalog number: 34080 )
- HEPES Sodium Salt (Thermo Fisher Scientific, Fisher BioReagents, catalog number: BP410-500 )
- Potassium chloride (KCl) (Sigma-Aldrich, catalog number: 793590-500 g )
- Tween® 20 (enzyme grade) (Thermo Fisher Scientific, Fisher BioReagents, catalog number: BP337-500 )
- 2-mercaptoethanol (Thermo Fisher Scientific, Fisher BioReagents, catalog number: BP176-100 )
- EDTA disodium salt (Thermo Fisher Scientific, Fisher BioReagents, catalog number: BP120-500 )
- ZnSO4.7H2O (Sigma-Aldrich, catalog number: Z4750-100 g )
- 5x HEPES/KCl (see Recipes)
- Microscale Thermophoresis Buffer (MST) buffer (see Recipes)
- MST with Zn2+ (see Recipes)
- MST with EDTA (see Recipes)
Equipment
- Multichannel pipettor (20-300 µl) (Gilson, model: FA10016 )
- Microplate reader with absorbance and chemiluminescent capacity (BioTek Instruments, model: Synergy 2 )
- Benchtop microplate shaker (IKA® Works, model: MS 3 digital )
Procedure
In this experiment, we have defined the His-tagged cognate binding partner as the immobilized ligand (S6-His) that interacts with two competing proteins, which are not immobilized (S18-1 or S18-2). Either of the two competing binding partners could be used to displace the other. However, we had antibodies only against the S18-2 protein and therefore the displacement protocol described here allows for detection of the S18-2 protein remaining bound to the S6 protein after displacement with the S18-1 protein. In general, if proteins binds to the same binding site with similar on and off rates, the order of addition of the competing proteins should not matter. Before proceeding with the experiment, there are two controls that need to be established. First, each competing protein should be added to separate wells without the presence of the His-tagged cognate protein. We did this control to confirm complete removal of the His-tag and to confirm the competing protein did not bind to the plate without the presence of S6-His. The second control is to determine the molar amount of the His-tagged protein that gives a linear range of detection, before the Ni2+-NTA binding sites become saturated. The plates used in this assay are 96 well Ni2+-NTA coated plates with a reported binding capacity of 10-20 pmol/well. We tested a range of His-tagged protein amounts from 5-150 pmol/well and found that the signal was saturated at 80 pmol when using 200 µl volume per well. Therefore, we used less than 80 pmol/well of the S6-His protein for the assay to ensure the molar concentrations of the competing untagged S18 proteins would be in excess of the His-tagged protein bound to the plate. Always use freshly purified and refolded proteins (or thawed aliquots stored at -80 °C) and ice-cold buffer for all dilutions and washes to maintain integrity of the ribosomal proteins throughout the assay.
- To begin this experiment purified recombinant proteins are needed. If His-tag is used for purification, the competing proteins, as in case of S18-1 and S18-2, must have their tags removed while the cognate protein/ligand, i.e. S6 protein, must remain His-tagged. As described previously (Prisic et al., 2015), we used hexa-histidine tags for S18-1, S18-2, and S6 and purified the recombinant proteins on cobalt columns under denaturing conditions and then refolded by dialysis. Using an engineered TEV cleavage site, we removed the histidine tag from S18-1 and S18-2 with TEV protease, followed by purification of the cleaved proteins with a His-tag purification resin matrix.
- Ensure the proteins are all in the same buffer; in this case MST buffer was used. If they are not in the same buffer, they should be re-dialyzed or buffer should be exchanged using gel filtration. The ribosomal proteins were refolded and stored in MST buffer, which was optimized for binding studies using microscale electrophoresis (Prisic et al., 2015). Considering that we observed strong interaction between S6 and each S18 protein in this buffer, we decided to use the same conditions in the displacement assay. Components did not interfere with Ni2+-NTA-His tag binding and had acceptable effect on ELISA, i.e. binding of primary and secondary antibodies, and HRP activity. MST buffer was used for all dilutions, including antibody dilutions, as well as washing steps. We did this assay in parallel, one with proteins dialyzed with Zn2+ and one with proteins dialyzed with EDTA. Therefore we would use MST with Zn2+ (10 µM ZnSO4) when working with Zn2+-dialyzed proteins or MST with EDTA (100 µM EDTA) when working with EDTA-dialyzed proteins.
- Determine the concentration of the proteins. For the ribosomal proteins, we used BioRad protein concentration assay, a modified Bradford method. BSA standards were prepared in MST buffer and protein concentration was determined from the standard curve when measuring the absorbance at 595 nm in a microplate reader. Molecular weights of the proteins were 11.9 kDa for S6-His, 9.6 kDa for S18-1 and 9.8 kDa for S18-2. The molar concentration of the proteins was determined from the protein concentration given from the Bradford assay using the following formula:
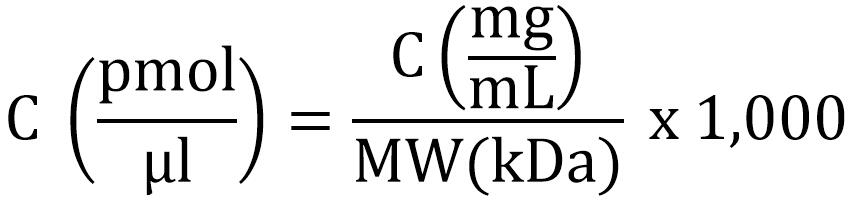
- Make a master mix of His-tagged protein diluted in MST for all wells to be tested. We used 30 pmol/well of the His-tagged protein and 200 µl well volumes, so the protein will be diluted to a final concentration of 150 µM. Aliquot 200 µl of diluted His-tagged protein into each well (Figure 1a). It is recommended that at least two to three technical replicates are done in parallel.
- Incubate plate for 30 min at room temperature with shaking at 300 rpm on a bench top plate shaker.
- Discard the solution from the plate and wash the wells four times with 200 µl MST. Washes are done with ice-cold buffer but are carried out at room temperature. Wells are soaked with wash buffer for 30-60 sec without shaking and tapped dry onto paper towels between each wash.
- Make a master mix of the S6 binding partner (i.e. S18-2 protein) at a molar concentration at least 1.5 times greater than that used for the His-tagged protein, so there is enough material to ensure complete saturation of the His-tagged protein. In our case, we used a molar concentration of 270 µM (54 pmol/well) of the S18-2 protein. Aliquot 200 µl of the master mix per well (Figure 1b).
- Incubate plate for 30 min at room temperature with shaking (300 rpm) on a bench top plate shaker.
- Discard the solution from the plate and wash as done in step 6.
- Prepare a range of molar concentrations for another S6-binding protein (i.e. S18-1) in the following manner. Prepare a solution at 4x the amount of the first S6 binding partner used in step 7 (in our case this is 1.08 mM). Create serial dilutions decreasing the concentration by half each step until 0.25x is achieved (67.5 µM).
- Aliquot 200 µl of each dilution per well. Include one well to which no competing protein is added to establish the full strength signal obtained from saturation of the His-tagged cognate protein without displacement (Figure 1c).
- Incubate plate for 30 min at room temperature with shaking (300 rpm) on a bench top plate shaker.
- Discard the solution from the plate and wash as done in step 6.
- Prepare the primary antibody (antibody specific to one of the competing proteins, in our case it was against S18-2) at 1:1,000 dilution in MST with 3% BSA. Aliquot 200 µl of the antibody solution to each well.
- Incubate plate for 30 min at room temperature with shaking (300 rpm) on a bench top plate shaker.
- Discard the solution from the plate and wash as done in step 6.
- Prepare the secondary antibody (conjugated to HRP) at 1:5,000 dilution in MST with 3% BSA. Aliquot 200 µl of the secondary antibody solution into each well, including controls.
- Incubate plate for 30 min at room temperature with shaking (300 rpm) on a bench top plate shaker.
- Discard the solution from the plate and wash as done in step 6.
- Using a multichannel pipettor, aliquot 200 µl of chemiluminescent substrate (e.g. 1:1 mix of SuperSignal Pico stable peroxide and substrate) to each well. Attempt to add substrate to each row of the plate (in our case each row was one complete replicate) in rapid succession, as the signal increases rapidly with time. Using a trough with premixed substrate will allow minimal rounds of pipetting. It is most important that each row of the plate has substrate added simultaneously using the multichannel pipette so that standardization of the well without competing protein and the treatment wells with competition for each replicate have substrate added simultaneously.
- Gently shake the plate for 1 min and read chemiluminescent signal (LUM) in a plate reader immediately (Figure 1d).
- Normalize the signal for each well to the signal obtained from the well in which no competing protein was added, representing a fully saturated signal. The signal obtained from each treatment well will be expressed as a percentage of the full strength signal obtained without competition (Figure 1e).
Representative data
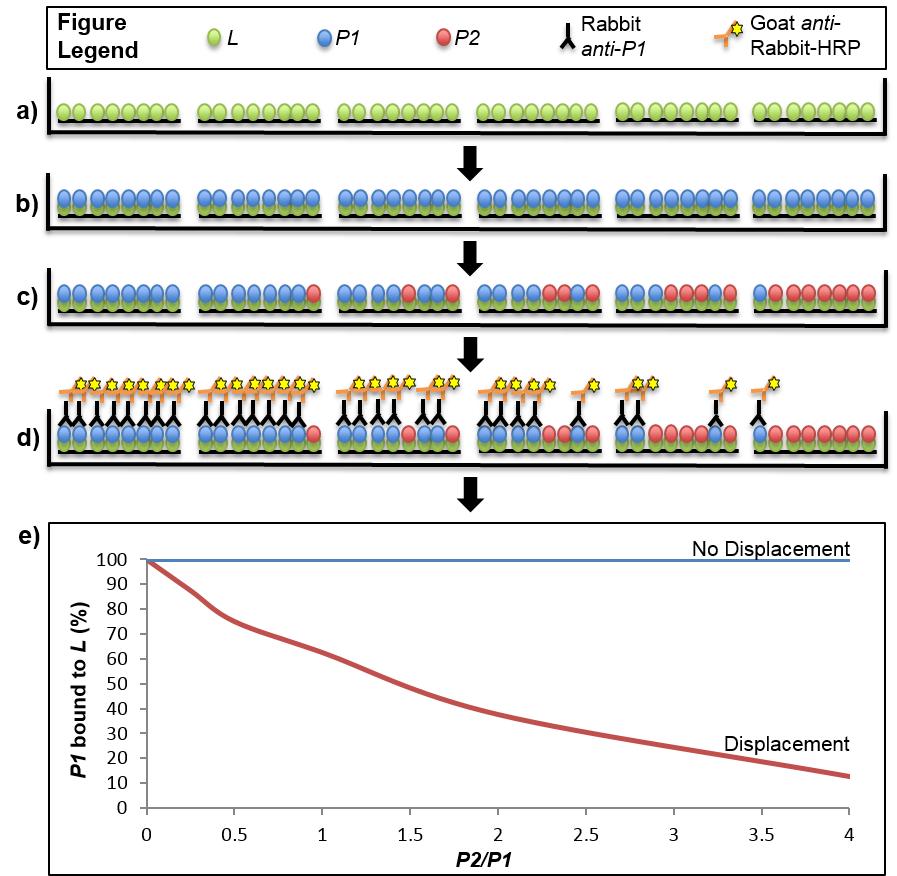
Figure 1. Stepwise depiction of a displacement-based ELISA. Virtual cross-sections through six wells of a 96-well plate are depicted throughout stages of the displacement assay. a). The ligand (L) is immobilized on a Ni2+-NTA plate via His-tag. b). The first binding partner, P1, is added in excess to saturate the binding sites offered by the immobilized ligand. c). The second binding partner, P2, is added to the wells in increasing amounts from 0-4X of the maximal theoretical concentration of P1 bound to L. In the event that displacement of P1 by P2 occurs, some of the P1 that was originally bound to L will be replaced with P2. The amount of P1 that is displaced will be dependent on the ratio of P2/P1 that was added to the well, with higher ratios leading to less P1 that remains bound with L. d). The result of the displacement assay is obtained through an ELISA with primary antibodies specific to P1 and secondary antibodies conjugated to horseradish peroxidase (HRP). Light emitted from a luminescent HRP substrate is P1 concentration-dependent and displacement is determined through normalization of the signal from each well to one in which no P2 was introduced (i.e. full saturation of L with P1). e). Displacement can be visualized by plotting the normalized signal of P1 across the tested range of P2/P1 ratios. The graph shows two hypothetical results where displacement or no displacement occurred. In the scenario described in the protocol P1 is S18-2, P2 is S18-1 and L is S6-His.
Recipes
- 5x HEPES/KCl (1 L)
123 g KCl
23.83 g HEPES Sodium Salt
900 ml H2O
Adjust pH to 7.6 with NaOH
Bring final volume to 1 L - MST buffer (20 mM HEPES pH 7.6, 330 mM KCl, 0.05% Tween 20, 14 mM 2-mercaptoethanol) (1 L)
200 ml 5x HEPES/KCl
2.5 ml 20% Tween® 20
1 ml 2-mercaptoethanol
Bring final volume to 1 L
Note: Always make MST buffer fresh and use the same day. - MST with Zn2+
MST buffer + 10 µM ZnSO4 - MST with EDTA
MST buffer + 100 µM EDTA
Acknowledgments
This work is supported by startup funds from University of Hawaii at Manoa.
References
- Prisic, S., Hwang, H., Dow, A., Barnaby, O., Pan, T. S., Lonzanida, J. A., Chazin, W. J., Steen, H. and Husson, R. N. (2015). Zinc regulates a switch between primary and alternative S18 ribosomal proteins in Mycobacterium tuberculosis. Mol Microbiol 97(2): 263-280.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Dow, A. and Prisic, S. (2016). Displacement-based ELISA: Quantifying Competition between Two Binding Partners for Interaction with a His-tagged Ligand Immobilized on a Ni2+-NTA Plate. Bio-protocol 6(5): e1745. DOI: 10.21769/BioProtoc.1745.
Category
Microbiology > Microbial biochemistry > Protein > Interaction
Biochemistry > Protein > Interaction > Protein-ligand interaction
Biochemistry > Protein > Interaction > Protein-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.