- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Super-resolution Imaging of Live BY2 Cells Using 3D-structured Illumination Microscopy
Published: Vol 6, Iss 1, Jan 5, 2016 DOI: 10.21769/BioProtoc.1697 Views: 9647
Reviewed by: Fanglian HeAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Aug 2015

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Light microscopy is the standard tool for studying sub-cellular structures however, owing to the diffractive properties of light, resolution is limited to 200 nm. Super-resolution microscopy methods circumvent this limit, offering greater resolution, particularly when studying fluorescently labeled sub-cellular structures. Super-resolution methods such as 3D-SIM (Structured Illumination Microscopy) fill a useful niche between confocal and electron microscopy. We have previously had success using fixed plant tissue samples with 3D-SIM (Bell and Oparka, 2014). However, sensitive structures can be altered by fixation and embedding procedures, so we developed a method for imaging live cells. In this protocol we used 3D-SIM to image the ER and Hechtian Strands in live, plasmolysed BY2 cells.
Materials and Reagents
- Microscope slides (Thermo Fisher Scientific)
- #1.5 coverslips (0.17 mm thick) (Thermo Fisher Scientific)
- BY2 cells expressing fluorescent marker
- 250 ml Erlenmeyer flasks (Thermo Fisher Scientific)
- Murashige and Skoog (MS) basal salt media (Sigma-Aldrich, catalog number: M5519 )
- Sucrose (Thermo Fisher Scientific)
- (2, 4-Dichlorophenoxy) acetic acid sodium salt monohydrate (Sigma-Aldrich, catalog number: D6679 )
- Calcofluor white/Fluorescent Brightener 28 (Sigma-Aldrich, catalog number: F3543 )
- 1 M D-Mannitol pure (Scientific Laboratory Supplies, catalog number: CHE1796 )
- Nail varnish
- BY2 growth media (see Recipes)
- Calcofluor White Stock solution (see Recipes)
Equipment
- Controlled temperature (28 °C) incubator or room
- Orbital shaker
- PersonalDV Deltavision Epi-fluorescence Inverted Microscope (GE Healthcare, Dharmacon)
- 3D-SIM microscope [DeltaVision OMX Blaze (GE Healthcare, Dharmacon) fitted with an Olympus PlanApo N 100x 1.42 NA oil objective]
- Edge sCMOS camera (PCO AG)
Software
- SoftWoRx 6.0 (GE)
Procedure
- Sample preparation
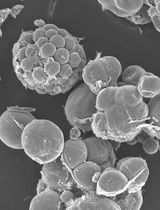
- Culture 40 ml of BY2 cell lines stably expressing a fluorescent reporter in 250 ml Erlenmeyer flasks with sterile Murashige and Skoog Basal Salts media supplemented with 3% sucrose and 2 μg ml-1 2,4-Dichlorophenoxyacetic acid. Grow cultures at 28 °C, in the dark, on an orbital shaker at 140 rpm. Aseptically sub-culture cells to fresh media weekly using a dilution of 1 ml of cells to 40 ml of media. Healthy cultures are pale yellow to light beige in colour and there should be sufficient growth that the cell suspension is more viscous than the media (Figure 1).
- At 4 days post-subculture, pipette 1 ml of cells into an Eppendorf tube and then stain cell walls (if required) with a final concentration of 3.5 μg ml-1 Calcofluor white for 5 min at room temperature. Excess stain is removed by allowing the cells to settle to the bottom of the tube and exchanging the media for fresh, twice.
- Plasmolyse cells by removing 450 μl of media and replacing it with 450 μl of 1 M Mannitol.
Note: Plasmolysis is not required, but enabled us to observe plasmodesmata more accurately. - Incubate at room temperature for 10 min, gently inverting the tube every 2 min to keep cells in suspension and then pipette 40 μl of cells onto a microscope slide. Lay a rectangular coverslip gently on top. Remove excess media by gentle pressure using a folded flat piece of absorbent tissue paper.
Note: It is important not to damage the cells, so the pressure must be light. - Seal all edges of the coverslip with nail varnish and begin imaging as soon as the varnish has dried.
- Locate candidate cells for imaging using a PersonalDV DeltaVision live-cell imaging system, which has stage coordinates mapped to the OMX. Use low level bright field illumination and mark the position on the slide using point visiting tool.
- Expose all marked cells to the appropriate wavelength of fluorescent light to check the calcofluor stain (abs350 nm/em455 nm) has worked well and that the cells are expressing a good level of fluorescent reporter (Figure 2). It is vital to keep this exposure as brief as possible in order to minimize bleaching prior to 3D-SIM imaging. The point visiting function allows the marked cells to be found swiftly on the OMX, thus avoiding unnecessary exposure to light.
- Culture 40 ml of BY2 cell lines stably expressing a fluorescent reporter in 250 ml Erlenmeyer flasks with sterile Murashige and Skoog Basal Salts media supplemented with 3% sucrose and 2 μg ml-1 2,4-Dichlorophenoxyacetic acid. Grow cultures at 28 °C, in the dark, on an orbital shaker at 140 rpm. Aseptically sub-culture cells to fresh media weekly using a dilution of 1 ml of cells to 40 ml of media. Healthy cultures are pale yellow to light beige in colour and there should be sufficient growth that the cell suspension is more viscous than the media (Figure 1).
- 3D-SIM Imaging
- We acquire 3D-SIM images using a DeltaVision OMX Blaze fitted with an Olympus PlanApo N 100x 1.42 NA oil objective. The 3D-SIM imaging protocol is based on that described in Schermelleh et al. (2010). Place the slide on the OMX and apply immersion oil. To minimise spherical aberration and optimize illumination modulation contrast, adjust the type of immersion oil to match the Refractive Index (RI) of the sample and imaging depth. Ideally, BY2 cells would be mounted in glycerol or similar to maintain the RI of the oil immersion objective but to keep the ER in good condition the cells must be mounted in media. Cells also need to be as close to the coverslip as possible.
- Find the selected cells using the Point List and then the Spiral Mosaic function to center the cell in the image. Determine top and bottom limits of z-stack quickly and efficiently, continuing to minimize the cell’s exposure to light.
- In the OMX 3D-SIM system, light from solid state lasers (405, 488 and 564 nm), shuttered by high speed tilt mirrors and coupled into a broadband single mode optical fiber is split into three beams. 3D-interference patterns in the sample plane are generated by focusing the beams onto the back focal plane of the objective lens. Striped illumination patterns are shifted by five phase steps and rotated by 3 angles (-60°, 0° and +60°), providing a set of 15 images per unprocessed z-section. Interference patterns are phase shifted by directing the outer two beams through a separate pair of windows with individual tilt control. Phase of the interference pattern at the sample plane is shifted due to the change in the path length for the respective outer beam, while lateral refractive beam translation is canceled by tilting a given window pair in complementary directions. Angles of pattern orientation are shifted by a tilt mirror, directing the three beams pattern to one of three mirror clusters; the beam pattern from each of the three rotation paths is redirected back to a common exit path by reflecting a second time from the tilt mirror. For descriptive diagrams see http://microscopy.lifesci.dundee.ac.uk/omx/omx.html.
- Select the lowest possible laser power and exposure times for each channel to minimize photo bleaching. Adjust exposure times similarly, typically between 100 and 200 ms, and also adjust the power of each laser to achieve optimal intensities of between 1,000 and 3,000 counts in a raw image acquired by a 15-bit dynamic range Edge sCMOS camera. Acquire image stack.
- Unprocessed image stacks are composed of 15 images per z-section (five phase-shifted images per each of three interference pattern angles). The microscope must be routinely calibrated by measuring channel specific optical transfer functions (OTFs) to optimize both lateral and axial image resolution.
- Adjust images from the different color channels, recorded on separate cameras, with the SoftWorx 6.0 alignment tool, based on alignment parameters obtained from calibration measurements with 100 nm-diameter TetraSpeck beads. Reconstruct super-resolution 3D image stacks with SoftWoRx 6.0 using channel specific OTFs and Wiener filter setting of 0.002.
- We acquire 3D-SIM images using a DeltaVision OMX Blaze fitted with an Olympus PlanApo N 100x 1.42 NA oil objective. The 3D-SIM imaging protocol is based on that described in Schermelleh et al. (2010). Place the slide on the OMX and apply immersion oil. To minimise spherical aberration and optimize illumination modulation contrast, adjust the type of immersion oil to match the Refractive Index (RI) of the sample and imaging depth. Ideally, BY2 cells would be mounted in glycerol or similar to maintain the RI of the oil immersion objective but to keep the ER in good condition the cells must be mounted in media. Cells also need to be as close to the coverslip as possible.
Representative data

Figure 1. Healthy 4-day old BY2 cell culture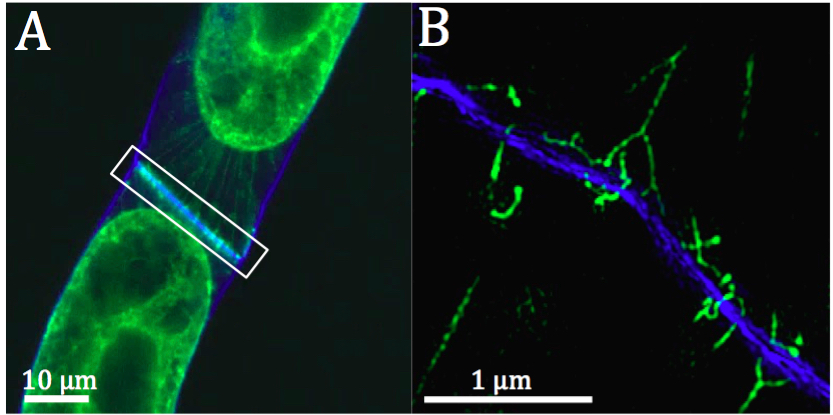
Figure 2. Representative image showing the resolution obtainable with 3D-SIM using plasmolysed live BY2 cells. A. Standard confocal image of BY2 cells expressing RTN6:GFP (green) and the cell wall stained with Calcofluor White (blue). B. Pseudo-coloured 3D-SIM image of the boxed region in A showing high resolution of the Hechtian strands at high magnification.
Notes
A 3D-SIM microscope takes considerable expertise to operate, with different manufacturers using bespoke software specifically designed for their hardware. Therefore, the 3D-SIM imaging part of this protocol is provided as a guide only. Each experiment will take considerable optimization of all 3D-SIM parameters as outlined in the protocol.
Recipes
- BY2 growth media
0.43% MS Basal Salts media
3% sucrose
2 μg ml-1 2, 4-Dichlorophenoxyacetic acid
ddH2O
Sterilize the media by autoclaving - Calcofluor White Stock solution
170 μg ml-1 Calcofluor white
Dissolve in ethanol
Stored in the dark at -20 °C
Acknowledgments
Use of the Deltavision OMX Blaze microscope at Dundee University was supported by an MRC Next Generation Optical Microscopy Award (Ref: MR/K015869/1). Development of this protocol was part of a project funded by grant BB/J004987/1 from the British Biotechnology and Biological Sciences Research Council (BBSRC) to Karl Oparka. We are grateful for the expert assistance of Dr. Markus Posch. Elements of this protocol have been adapted from those previously described in Bell and Oparka (2015) and Knox et al. (2015).
References
- Bell, K. and Oparka, K. (2015). Preparative methods for imaging plasmodesmata at super-resolution. Methods Mol Biol 1217: 67-79.
- Knox, K., Wang, P., Kriechbaumer, V., Tilsner, J., Frigerio, L., Sparkes, I., Hawes, C. and Oparka, K. (2015). Putting the squeeze on Plasmodesmata: A role for reticulons in primary plasmodesmata formation. Plant Physiol 168(4): 1563-1572.
- Schermelleh, L., Heintzmann, R. and Leonhardt, H. (2010). A guide to super-resolution fluorescence microscopy. J Cell Biol 190(2): 165-175.
Article Information
Copyright
© 2016 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Bell, K., Oparka, K. and Knox, K. (2016). Super-resolution Imaging of Live BY2 Cells Using 3D-structured Illumination Microscopy. Bio-protocol 6(1): e1697. DOI: 10.21769/BioProtoc.1697.
- Knox, K., Wang, P., Kriechbaumer, V., Tilsner, J., Frigerio, L., Sparkes, I., Hawes, C. and Oparka, K. (2015). Putting the squeeze on Plasmodesmata: A role for reticulons in primary plasmodesmata formation. Plant Physiol 168(4): 1563-1572.
Category
Plant Science > Plant cell biology > Cell imaging
Plant Science > Plant cell biology > Cell structure
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.