- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Analysis of Developing Pollen Grains within Intact Arabidopsis thaliana Anthers by Olympus Two-Photon Laser Scanning Microscopy


Published: Vol 5, Iss 23, Dec 5, 2015 DOI: 10.21769/BioProtoc.1677 Views: 10413
Reviewed by: Tie LiuNing LiuAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Nov 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








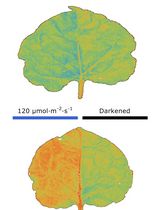
Abstract
The method consists of imaging developing pollen grains as they form within intact, immature Arabidopsis thaliana anthers. Using two-photon excitation in the infrared wavelength range, the intrinsic fluorescence (autofluorescence) of developing pollen grains and surrounding sporophytic tissues of the anther wall, including the tapetum, middle layer, endothecium and epidermis, can be visualized in the three-dimensional space of an intact anther. In contrast to conventional confocal microscopy, the application of red-shifted light by two-photon microscopy improves depth penetration into specimens, while the scattering of light and subsequent phototoxicity is minimized, making this a superior method for imaging the developing pollen grains and tapetal cells enclosed within anthers. The technique described was optimized for the detection of autofluorescent components of the pollen wall, including sporopollenin and the pollen coat, and provided spatial and developmental data on the autofluorescent metabolites in anthers of wild-type and pollen wall mutant plants (Quilichini et al., 2014). The use of two-photon imaging of live, intact anthers holds potential for future studies aimed at understanding the spatial relationship between gametophytic and sporophytic tissues during pollen development and the distribution of metabolites or fluorescently-tagged proteins within developing anthers.
Materials and Reagents
- Ruler with millimetre divisions
- Glass bottom Petri dishes (MatTek Corporation, catalog number: P35G-1.5-14C )
- Coverslips (No.1.5, 0.17 mm thick, 22 x 22) (Thermo Fisher Scientific, catalog number 12-541B )
- Transfer pipettes 1.5 ml
- Arabidopsis thaliana plants in the flowering stage
- Non-toxic food-grade paraffin wax maintained in a liquid state at 40 ºC in a paraffin bath (Thera-Band, catalog number: 24050 )
- Distilled water
Equipment
- Spectra-Physics MaiTai HP Titanium:Sapphire mode-locked pulsed laser, with tuneable wavelengths spanning 690 to 1,040 nm.
- Olympus Fluoview 1000 scan head (Olympus America Inc., model: FV1000MPE ) modified by Olympus for two-photon imaging by the inclusion of a light path for the pulsed laser input and the housing for the non-descanned, multialkali side window photomultiplier tubes (PMTs).
- Olympus BX61WI upright microscope
- Olympus multiphoton microscope filter cube (440/40 bandpass filter) (OLYMPUS, model: FV10-MRVGR/XR 4CH NDD FILTER )
- Olympus multiphoton microscope dedicated objective lens optimized for transmitting IR light (25x, 1.05 numerical aperture, 2.0 mm working distance) (OLYMPUS, model: FV10-SNPXLU )
- Light tight, custom made multiphoton microscope enclosure. To build an enclosure, aluminum extruded components (Item North America) and flat-black painted fibre board can be used
- Forceps for fine dissections (Electron Microscopy Sciences, catalog number: 72700-D )
- Olympus stereomicroscope (magnification range of 6.3-63x) (OLYMPUS, model: SZX10 )
Software
- Olympus FluoView FV10-ASW 3.01
- Spectra-Physics MaiTai Control laser software
- Volocity version 6.1.1 software package
Note: ImageJ 1.47v can also be used to process images.
Procedure
New users learning to operate an Olympus two-photon microscope or similar system should be supervised by an experienced user or technician. The protocol described herein is specific to the Olympus system and should only be used as a supplementary resource when operating other two-photon imaging platforms.
- Microscope set-up
- Operating the MaiTai laser.
Turn the laser power on, tune the laser to the desired wavelength and open the shutter using the Spectra-Physics MaiTai Control (see Figure 1). Although the laser can be operated through FluoView or separately through the Spectra-Physics MaiTai Control application, it is recommended to turn the laser on through the Spectra-Physics application, independently from the FluoView software, to prevent laser shut down in the case of software malfunctions.- Turning the laser on.
The laser should be set to 800 nm when not in operation (Figure 1, left panel) and should warm up at this wavelength (Figure 1, middle panel). Select and hold down ‘ON’ to turn the laser on (Figure 1, left panel). Wait for the laser to begin pulsing (indicated by the green box under ‘Pulsing’) and the IR Power level (indicated by a red bar) to stabilize (Figure 1, middle panel). Ensure that laser pulsing (mode-lock) status is reached. - Tuning the laser to the imaging wavelength.
After a 30 min laser warm-up, the infrared (IR) power level should be stabilized. Tune the laser set wavelength from 800 nm to 720 nm by entering the imaging wavelength ‘720’, ‘enter’ to tune the laser to this wavelength (Figure 1, right panel). Ensure the IR Power reaches 2.49 watts when using 720 nm and 3% laser power. Also ensure the correct 720 nm wavelength is entered in the Fluoview software Acquisition settings under Laser.
Note: As you change laser wavelength, IR Power also changes. To obtain consistent emission power at different wavelengths, the laser power must be adjusted each time the laser wavelength is changed. Enter the correct laser wavelength in FluoView, in the Acquisition Settings Panel under Laser. - Open the reflected light shutter.
Click on the closed (grey) shutter button to switch the shutter to the open (yellow) position (Figure 1). Manually open the shutter on the microscope unit, located beside the filter wheel.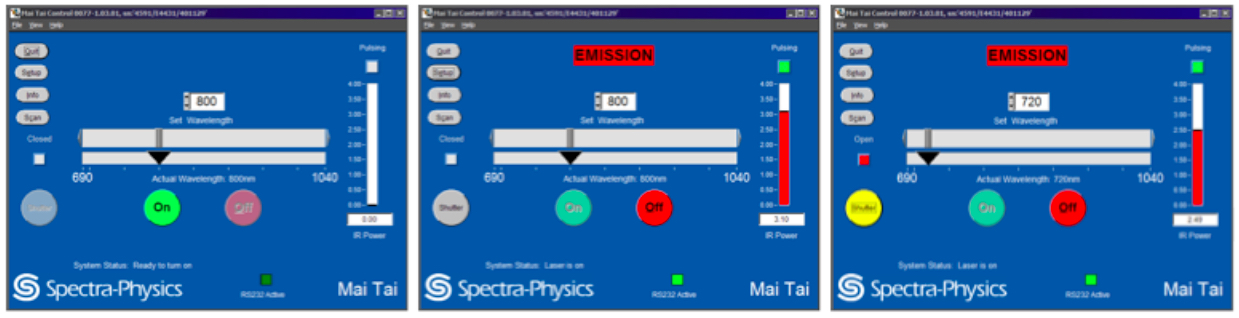
Figure 1. MaiTai laser operation through the Spectra-Physics Laser Control interface. From left to right: Adjusting laser parameters as shown turns the laser power on and tunes the laser to the desired wavelength. Starting with the left panel, the laser power is off and set to 800 nm by the previous user. Select and hold down ‘ON’ to turn on the laser on. Wait for the laser to begin pulsing (indicated by the green box under ‘Pulsing’) and the IR Power level (indicated by a red bar) to stabilize. After sufficient laser warm-up, tune the laser to the imaging wavelength of 720 nm. Ensure the IR Power level stabilizes. Click on the closed (grey) shutter button to switch the reflected light shutter to the open (yellow) position.
- Turning the laser on.
- Install the objective.
Select a clean objective and screw into position on the microscope. For the Olympus FV1000MPE multiphoton microscope, the XLPLN 25x water immersion objective is recommended, as it is optimized for multiphoton imaging (Figure 2A). Select the correct objective in the FluoView Acquisition Settings Panel, under Microscope. - Filter cube selection.
Select the Olympus BFP/GFP/RFP/Ds Red filter cube, with 440/40 bandpass filter (420-460 nm; Figure 2B). Always handle the filter cube with gloves. Screw the filter cube into place within the filter cube chamber located behind the filter wheel and sliders. - Adjusting objective lens and stage position.
The microscope stage can be adjusted in the x- and y-axes with the joystick control (Figure 2C). To move the objective lens in the z-axis, towards or away from the specimen, use the focus remote controller (Figure 2D).
Figure 2. Adjustable microscope hardware. A. The Olympus XLPlan N 25X W MP objective lens, recommended for multiphoton imaging. B. Olympus filter cube (FV10-MRVGR/XR 4CH NDD FILTER, BFP/GFP/RFP/Ds Red, 380-560 nm). C. Stage control joystick. D. Focus remote control. - Directing the light path.
For transmitted light viewing or two-photon imaging, different light path configurations are required. For transmitted light viewing, the sliders must be in the outward position to allow light to move to the oculars (Figure 3A) and the filter wheel should be in the DICT position (Figure 3C). For four-channel two-photon imaging, ensure both sliders are in (Figure 3B) and move the filter wheel to position 2 (Figure 3D). For the Olympus FV1000 MPE system, the R690 position on the filter wheel is meant for two-photon imaging and the DICT position allows transmitted light viewing.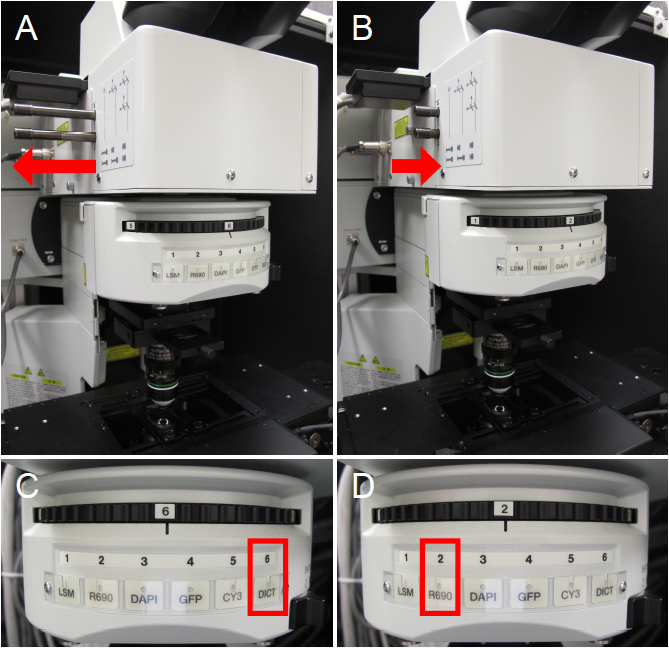
Figure 3. Microscope configurations. A. Microscope sliders are in the outward position for transmitted light viewing. B. Microscope sliders are in the inward position for two-photon imaging with emission capture in four channels. C. Filter wheel is in the DICT position for transmitted light viewing. D. Filter wheel is in the R690 position for two-photon imaging.
- Operating the MaiTai laser.
- Specimen preparation
- Select anthers in the free microspore stage of pollen development.
- With forceps, remove a floral cluster from your Arabidopsis plant of interest (Figure 4A). Discard open flowers.
- Under a dissecting microscope, separate each bud from the floral cluster using forceps (Figure 4B).
- For stamens in the free microspore stage, select buds measuring 0.7 to 1.2 mm from base to apex (Figure 4B, boxed in the middle row). Buds measuring <0.5 mm, 0.5-0.7 mm typically contain anthers in the microspore mother cell or tetrad stages of pollen development, respectively (Figure 4B, see bracketed floral bud cluster). Buds measuring >1.2 mm typically contain anthers in the late stages of pollen development, characterized by tricellular pollen and tapetum degeneration (Figure 4B, see bracketed bottom row of buds).
Note: The developmental stage of each stamen can vary between buds of the same length, among the six stamens within one bud and even occasionally between locules of the same anther. For these reasons, numerous anthers for each genotype of interest should be analyzed from multiple buds, to ensure the correct stage(s) of interest are identified. - Following bud size selection, carefully remove sepals and petals from one bud of interest (Figure 4C) and excise stamens into the microwell of a glass bottom Petri dish (Figure 4D). When handling each stamen with forceps, clasp the filament to avoid damage to the anther.
- Cover the stamens with distilled water. Gently remove all air bubbles trapped in the microwell. To remove air bubbles along the anther outer surface, hold each stamen underwater with forceps and gently manipulate bubbles away from the anther surface. Removing all air bubbles is critical, as air bubbles interfere with imaging. If air bubbles will not dissipate, the anther is not selected for imaging. Anthers ready for imaging should sink.
- Discard anthers with any visible damage due to dissections (such as the shrivelled anther with a detached filament, circled in Figure 4D). Anthers submerged in water (Figure 4E) but associated with air bubbles should be discarded.
- Seal your specimens under a coverslip with paraffin along all edges (Figure 4F).
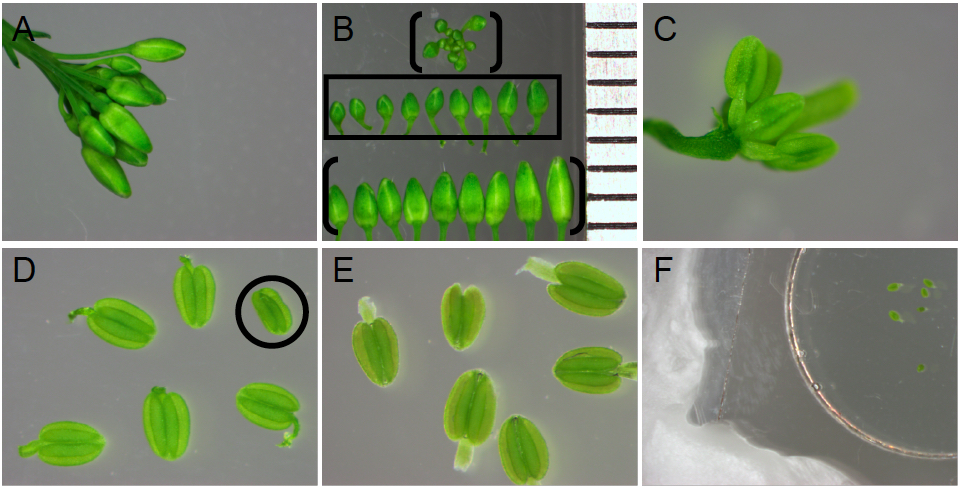
Figure 4. Stamen selection and preparation for imaging. A. Arabidopsis flower bud cluster with open flowers removed. B. Bud size selection. All buds displayed are from flower bud cluster in (A). Ruler on right edge depicts 1 mm intervals. Buds measuring 0.7-1.2 mm in length (middle row, boxed) are selected, as they contain anthers in the free microspore stages of pollen development before tapetum programmed cell death. Buds exceeding 1.2 mm in length (bracketed bottom row in panel B) or under 0.7 mm in length (in the floral cluster, bracketed in panel B) are discarded. C. Bud showing exposed stamens and carpel, after sepal and petal removal. D. Anthers after removal from flower pedicel but prior to submergence in water. Anthers with damage caused by dissections (such as the circled anther lacking a filament and appearing shrivelled) are discarded. E. Anthers submerged in distilled water, below paraffin sealed coverslip. Anthers associated with air bubbles are not imaged. F. A coverslip covering the microwell and submerged anthers is sealed to the Petri dish on all sides with paraffin.
- With forceps, remove a floral cluster from your Arabidopsis plant of interest (Figure 4A). Discard open flowers.
- Specimen loading and alignment.
Place a large water droplet onto the sample coverslip and lower objective lens until it is partially submerged. Bring the filter wheel to the DICT position for transmitted light. Turn the transmitted light lamp on (in FluoView under Image Acquisition Control) and pull both microscope sliders to the outward position to allow light to move to the oculars. Using the stage control joystick and the focus remote controller (Figure 2C) to centre your region of interest and bring your specimen into focus. Turn the transmitted light off and move the filter wheel to position 2 (R690) for two-photon imaging. - Enter the two-photon imaging mode.
In the FluoView program Image Acquisition Control panel, select the Dye List menu. Clear all dyes from the list before selecting ‘Two Photon’. - Check the light path.
For emission capture in the channels of interest, move the SDM570 dichroic mirror into the light path for four-channel imaging by sliding both microscope sliders inward. This configuration allows emission collection from channels 1-4 (RXD1-4, Figure 5). In the Quilichini et al. (2014) study, emissions for hydroxycinnamic acids and related compounds were collected in the low wavelength emission range of 420-460 nm (in RXD1), while chlorophylls and related compound emissions were collected in higher wavelengths, from 495-540 nm (in RXD2).
Note: The two-photon microscope system described in this protocol consists of a separate non-descanned light path with no pinhole in front of the PMTs. However, opening the pinhole for two-photon imaging on other systems with conventional confocal capabilities may be required.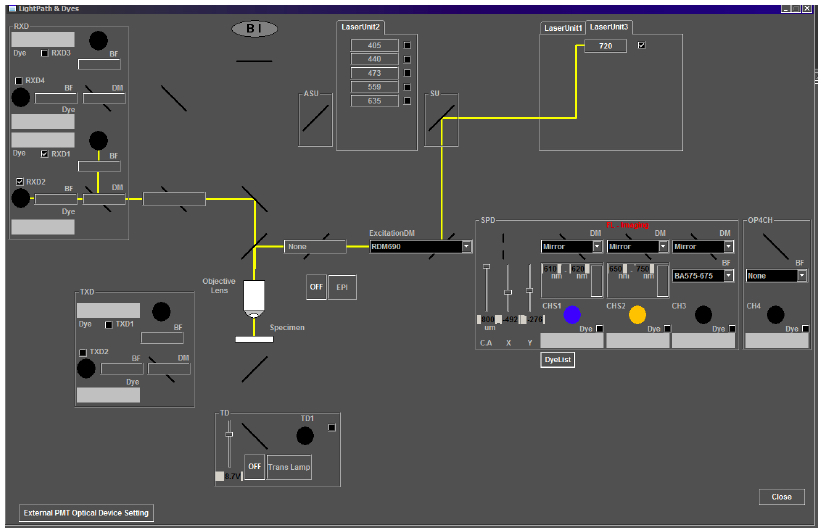
Figure 5. Light path for two-photon imaging on the Olympus FV1000MPE multiphoton microscope. For the correct light path for imaging anther autofluorescence, check that the laser path (depicted by a yellow line) in the Light Path and Dyes window of the Image Acquisition Control panel includes Laser Unit 3 at 720 nm, the excitation dichroic mirror, RDM690, and the correct emission capture channels, RXD1 and RXD2. - Image acquisition parameters.
In Fluoview, enable the two emission channels of interest by selecting RXD1 and RXD2 within the Image Acquisition control window. During scans and image collection, the live view window can be configured to display RXD1 and RXD2 emission, or an overlay of these channels. Different display pseudocolour options are available through the Live View window under LUT. The Hi-Lo display function is recommended, as it provides information on the intensity of emission over the imaging area. Using this display, overexposed pixels will appear red, while underexposed, dim pixels appear blue. Laser power and channel voltage settings, as detailed in this protocol below, were selected to maximize the signal intensity from the mixture of cell types within developing anthers, while avoiding sample rupture and pixel saturation. - Minimize sample damage.
During alignment, use a fast scan (called focus x 2 or focus x 4) to align your specimen without causing significant sample bleaching. Collect images using the xy scan function to collect a single frame image. To collect a z-stack, set the start and end z-positions for your sample of interest, typically surrounding one anther locule, with a 1 µm step size. Capture the z-stack by selecting depth (under the xy scan function) prior to clicking xy scan. - Minimize light pollution.
Detectors in the two-photon system are not protected by a pinhole, as in conventional confocal microscopy, to ensure all light in the focal volume is collected. To minimize light collection from external sources, ensure lighting in the room is minimized by sealing off the microscope within an opaque enclosure, performing imaging in a dark room, and operating the Fluoview program in the dark view. - Optimal imaging parameters for Arabidopsis anthers.
See table below for Acquisition Settings and Image Acquisition Controls. When different magnifications are used, it is recommended that the aspect ratio be adjusted accordingly, to keep pixel size as close to 0.2 µm/pixel as possible.Laser power 3% Laser wavelength (IR power) 720 nm (2.49 W) Scan mode XY Scan direction One-way Image size (Aspect ratio) 800 x 800 pixels (1:1) Zoom 3x Pixel size 0.212 µm/pixel Pixel dwell time 4.0 μs/pixel Z-stack step size 1.0 μm Integration type Kalman Line Averaging Integration count 3 RXD1 HV, Gain, Offset 650 V, 1, none RXD2 HV, Gain, Offset 650 V, 1, none - Correcting brightness at depth.
To improve emission capture from fluorophores located deep below the surface, either laser power or high voltage (HV, which controls the voltage applied on the PMTs, post-acquisition) can be adjusted for different z-positions (depths). This can be a useful tool, particularly for large specimens where valuable information is located at depth. Although this feature can be useful for imaging dim samples or locules positioned at great depths, it is time consuming, can cause specimen rupture if laser power settings are too high, and optimal settings over the z-series are generally sample specific. For consistency between genotypes and because anther tissues of interest are sufficiently bright without these adjustments, this correction was not applied for the imaging in our study (Quilichini et al., 2014). - Image processing.
Save each image or z-stack as an .oif file for processing in Volocity v 6.1.1, or import the image sequence into ImageJ equipped with the Bio-Formats plug-in (Abramoff et al., 2004; Linkert et al., 2010). Image processing commonly includes applying false-colour to the emission from each channel, overlaying channels into one merged image or stack, adjusting brightness and/or contrast in a consistent manner, adding scale bars, or creating a z-projection from a z-stack. - Instrument shutdown.
After imaging, return the laser to 800 nm prior to turning off laser power. Close the shutter on the microscope and through the software used to operate the laser. Move the objective to the uppermost position prior to closing the Fluoview program.
- Select anthers in the free microspore stage of pollen development.
Notes
Readers are encouraged to consult the Olympus Microscopy Resource Center (link below) for further information on the theory and applications of multiphoton fluorescence microscopy.
http://www.olympusmicro.com/primer/techniques/fluorescence/multiphoton/multiphotonintro.html.
Acknowledgments
We thank the University of British Columbia BioImaging Facility, particularly Kevin Hodgson, for training, technical assistance and discussions. This work was supported by Canadian Natural Sciences, Engineering Research Council (NSERC) Discovery Grants and the Working on Walls NSERC CREATE program. This protocol was developed for a study on pollen wall formation in Arabidopsis (Quilichini et al., 2014).
References
- Abramoff, M. D., Magalhaes, P. J. and Ram, S. J. (2004). Image Processing with ImageJ. Biophotonics International 11(7): 36-42.
- Linkert, M., Rueden, C. T., Allan, C., Burel, J. M., Moore, W., Patterson, A., Loranger, B., Moore, J., Neves, C., Macdonald, D., Tarkowska, A., Sticco, C., Hill, E., Rossner, M., Eliceiri, K. W. and Swedlow, J. R. (2010). Metadata matters: access to image data in the real world. J Cell Biol 189(5): 777-782.
- Olympus FV1000 MPE Microscope User Guide.
- Quilichini, T. D., Samuels, A. L. and Douglas, C. J. (2014). ABCG26-mediated polyketide trafficking and hydroxycinnamoyl spermidines contribute to pollen wall exine formation in Arabidopsis. Plant Cell 26(11): 4483-4498.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Quilichini, T. D., Samuels, A. L. and Douglas, C. J. (2015). Analysis of Developing Pollen Grains within Intact Arabidopsis thaliana Anthers by Olympus Two-Photon Laser Scanning Microscopy . Bio-protocol 5(23): e1677. DOI: 10.21769/BioProtoc.1677.
Category
Plant Science > Plant developmental biology > Morphogenesis
Plant Science > Plant cell biology > Cell imaging
Cell Biology > Cell imaging > Two-photon microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.