- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Telomere Restriction Fragment (TRF) Analysis


Published: Vol 5, Iss 22, Nov 20, 2015 DOI: 10.21769/BioProtoc.1658 Views: 22134
Reviewed by: HongLok LungVanesa Olivares-IllanaAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Jan 2015
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
While telomerase is expressed in ~90% of primary human tumors, most somatic tissue cells except transiently proliferating stem-like cells do not have detectable telomerase activity (Shay and Wright, 1996; Shay and Wright, 2001). Telomeres progressively shorten with each cell division in normal cells, including proliferating stem-like cells, due to the end replication (lagging strand synthesis) problem and other causes such as oxidative damage, therefore all somatic cells have limited cell proliferation capacity (Hayflick limit) (Hayflick and Moorhead, 1961; Olovnikov, 1973). The progressive telomere shortening eventually leads to growth arrest in normal cells, which is known as replicative senescence (Shay et al., 1991). Once telomerase is activated in cancer cells, telomere length is stabilized by the addition of TTAGGG repeats to the end of chromosomes, thus enabling the limitless continuation of cell division (Shay and Wright, 1996; Shay and Wright, 2001). Therefore, the link between aging and cancer can be partially explained by telomere biology. There are many rapid and convenient methods to study telomere biology such as Telomere Restriction Fragment (TRF), Telomere Repeat Amplification Protocol (TRAP) (Mender and Shay, 2015b) and Telomere dysfunction Induced Foci (TIF) analysis (Mender and Shay, 2015a). In this protocol paper we describe Telomere Restriction Fragment (TRF) analysis to determine average telomeric length of cells.
Telomeric length can be indirectly measured by a technique called Telomere Restriction Fragment analysis (TRF). This technique is a modified Southern blot, which measures the heterogeneous range of telomere lengths in a cell population using the length distribution of the terminal restriction fragments (Harley et al., 1990; Ouellette et al., 2000). This method can be used in eukaryotic cells. The description below focuses on the measurement of human cancer cells telomere length. The principle of this method relies on the lack of restriction enzyme recognition sites within TTAGGG tandem telomeric repeats, therefore digestion of genomic DNA, not telomeric DNA, with a combination of 6 base restriction endonucleases reduces genomic DNA size to less than 800 bp.
Materials and Reagents
- Whatman 3MM chromatography paper (46 x 57 cm) (Thermo Fisher Scientific, catalog number: 05-714-5 )
- 25 ml serological pipette (Thermo Fisher Scientific, catalog number: 13-668-2 )
- DNeasy Blood and Tissue Kit (QIAGEN, catalog number: 69504 )
- Proteinase K (QIAGEN, catalog number: 19131 or 19133 )
- Enzymes
- HhaI 20,000 units/ml(New England BioLabs, catalog number: R0139L )
- HinF1 10,000 units/ml (New England BioLabs, catalog number: R0155L )
- MspI 20,000 units/ml(New England BioLabs, catalog number: R0106S )
- HaeIII 10,000 units/ml (New England BioLabs, catalog number: R0108L )
- RsaI 10,000 units/ml(New England BioLabs, catalog number: R0167L )
- AluI 10,000 units/ml (New England BioLabs, catalog number: R0137L )
- NE Buffer2 10x concentrate (New England BioLabs, catalog number: B7002S )
- HhaI 20,000 units/ml(New England BioLabs, catalog number: R0139L )
- Uracil DNA Glycosylase (UDG) 5,000 units/ml (New England BioLabs, catalog number: M0280S )
- Klenow Fragment (3’→5’ exo-) 5,000 units/ml (New England BioLabs, catalog number: M0212S )
- DEPC-treated water (Life Technologies, catalog number: AM9906 )
Note: Currently, it is “Thermo Fisher Scientific, AmbionTM, catalog number: AM9906”. - Phosphate Buffered Saline (PBS) (Santa Cruz Biotechnology, ChemCruz, catalog number: sc-24947 )
- Tris-Acetate-EDTA (TAE) buffer (Thermo Fisher Scientific, catalog number: BP1332-1 )
- Tris-Base Ultrapure (Research Products International Corp., catalog number: T60040-5000.0 )
- Ethylenediamine Tetraacetic Acid (EDTA), Disodium Salt Dihydrate (Thermo Fisher Scientific, catalog number: BP120-1 )
- Boric acid (Sigma-Aldrich, catalog number: B6768 )
- UltraPureTM Agarose (Thermo Fisher Scientific, InvitrogenTM, catalog number: 16500-500 )
- GelRed Nucleic Acid Stain (PHENIX Research Products, catalog number: RGB-4102-1 )
- Radiolabelled TRF Marker (Herbert et al., 2003)
- DNA marker (Bionexus, catalog number: BN2050 )
- Sodium Chloride (NaCl) (Thermo Fisher Scientific, catalog number: S271-10 )
- Sodium Hydroxide (NaOH) (Thermo Fisher Scientific, catalog number: BP359-212 )
- Ficoll-Paque Plus (Thermo Fisher Scientific, catalog number: 45-001-749 )
- Polyvinylpyrrolidone (Sigma-Aldrich, catalog number: PVP40 )
- Bovine Serum Albumin (BSA), Fraction V (Gemini Bio-Products, catalog number: 700-106P )
- dCTP, [α-32P]-6,000 Ci/mmol 20 mCi/ml EasyTide Lead, 500 µCi (PerkinElmer, catalog number: NEG513Z500UC )
- 20x Saline-Sodium Citrate (SSC) (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15557-036 )
- Sodium Dodecyl Sulfate (SDS) (Sigma-Aldrich, catalog number: L4509 )
- 10x Buffer M (Roche Diagnostics, catalog number: 11417983001 )
Note: Currently, it is “Sigma-Aldrich, catalog number: 11417983001”. - 10x PBS buffer-phosphate buffer saline (see Recipes)
- 1x TAE buffer-Tris-Acetate-EDTA (see Recipes)
- 5x TBE buffer-Tris-Borate-EDTA (see Recipes)
- Hybridization solution (see Recipes)
- 100x Denhardt solution (see Recipes)
- 6x Glycerol and Bromophenol blue Loading Dye (see Recipes)
Equipment
- Water bath (Thermo Fisher Scientific, model: Isotemp 205 )
- Microcentrifuge (Eppendorf, model: 5424 )
- Nanodrop (Thermo Fisher Scientific, Nanodrop Technologies, model: ND-1000 UV/Vis Spectrophotometer )
- Gel Tank (Thermo Fisher Scientific, model: OwlTM A2 Large Gel Systems for 20 x 25 cm gel size )
- Savant Slab Gel dryer (SibGene, model: SGD4050 )
- Hybridizer (Bibby Scientific, Techne, model: Hybrigene )
- Cylinder (Bibby Scientific, Techne, model: FHB16/FHB15 )
- Thermocycler (LABGENE Scientific, Biometra, model: T1 Thermoblock )
- G-BOX (Syngene, model: G-BOX F3 )
- Power supply (Whatman Biometra, model: 250 EX )
- Microwave oven (GE, model: 1540WW002 )
- Screen (Molecular Dynamics, model: Kodak Storage Phosphor Screen )
- Typhoon PhosphorImager scanner system (Amersham Biosciences, GE Healthcare, model: Typhoon TRIO )
Software
- Image Quant® software (Molecular Dynamics)
- Graph Pad Prism 6®
Procedure
- Cell pellet: Prepare cell pellets (1 x 106 - 2 x 106) in 2 ml polypropylene screw-cap tubes. After removing the supernatant, cell pellets can be frozen at -80 °C.
Note: It is preferred to wash cell pellet with 1x PBS, but this step is not required because the small amount of medium in the cell pellet does not interfere with subsequent steps. - Cell lysis: Re-suspend the cells that are fresh or just thawed on ice from -80 °C with 200 μl 1x PBS and add 20 μl proteinase K (20 mg/ml).
- DNA extraction: Use DNeasy Blood and Tissue Kit to get high DNA yield. Quantify DNA samples by Nanodrop.
Note: Multiple methods are available to extract DNA from cells. We prefer the commercial kit since it can easily be performed to get high yield DNA. - DNA digestion: Digest 2.5 μg DNA for 4 h to overnight at 37 °C with combination of restriction enzymes (HhaI, HinF1, MspI, HaeIII, RsaI, AluI).
Prepare 2.5 μg DNA in dH2O to make 40 μl total volume.
Add 10 μl enzyme digestion mix to DNA sample (40 μl), so total volume is 50 μl. Incubate in a water bath or heating block overnight at 37 °C.
Enzyme digestion mix:
1 sample HhaI 0.25 μl HinF1 0.5 μl MspI 0.25 μl HaeIII 0.5 μl RsaI 0.5 μl AluI 0.5 μl NE buffer 2 5.0 μl H2O 2.5 μl Total: 10 μl - Gel migration: Load radiolabeled TRF ladder and unlabeled molecular weight marker on either or both sides of the samples. Separate digested DNA on 0.7% (w/v) agarose gel stained with gel red (1/20,000x) for 18 h at 70 volts for a 25 cm long gel in 1x TAE or 0.5x TBE buffer. 18 h after samples run, check the gel under a UV source (e.g., at G-BOX) to make sure genomic DNA is completely digested and runs as a smear below the 800 bp molecular weight marker (Figure 1A). Anything above 800 bp shows incomplete genomic DNA digestion and this could interfere with the telomere signal (Figure 1B).
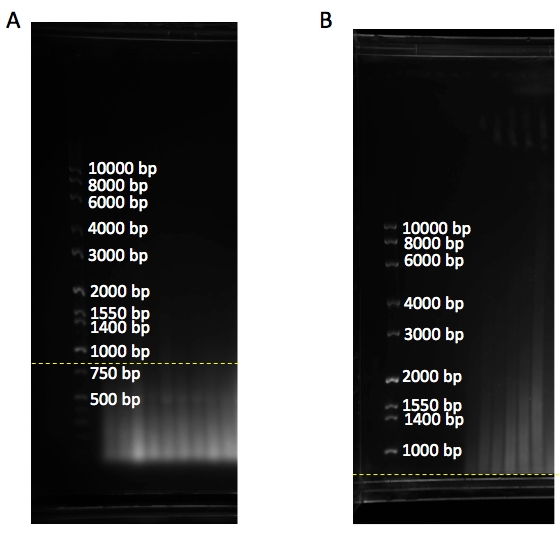
Figure 1. Gel with DNA ladder marker. A. DNA samples run as a smear below 800 bp (yellow dashline), showing complete digestion of genomic DNA. B. DNA samples are above 800 bp (yellow dashline), showing incomplete genomic digestion. bp: base pair
Notes:- Add 5 μl loading dye to the samples. While 0.5x TBE can be used to analyze less than ~8 kb, 1x TAE buffer can be used for the ones that have longer telomeres (8 to 20 kb range) to have better separation.
- 400 ml volume is good enough for 0.7% (w/v) agarose gel in large gel system.
- Radiolabeled TRF marker can be visualized after hybridization with telomere sequence-specific probe. The unlabeled, digested plasmid DNA can be visualized with Gel Red, not with the telomere sequence specific probe.
- To prevent leaking of the gel from the gel tray: Wait 20 min after agarose is dissolved in microwave. During this time, seal the space between the gel tray and gaskets (edges of gel tray) with 5-10 ml agarose gel (Figure 2A). Wait 30 min following the pour of the gel (do not forget the put comb when you pour the gel).
- Higher concentration of agarose in buffer will cause poor separation of samples and TRF marker (Figure 2B) and also processing gel drying will take a much more time than lower concentration of agarose.
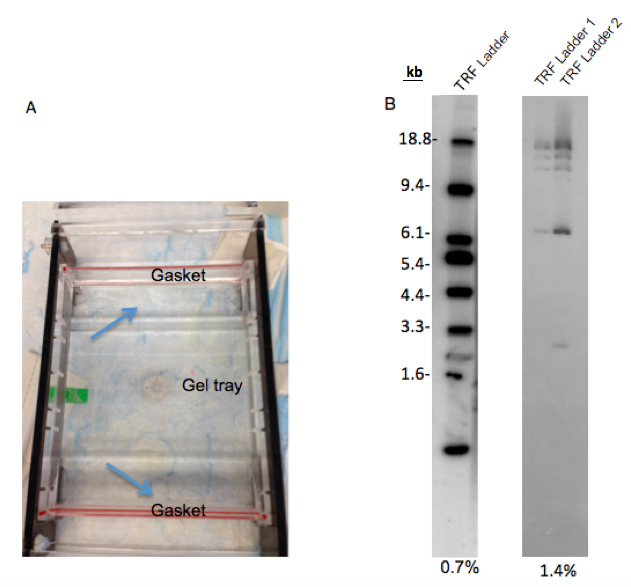
Figure 2. Preparation of agarose gel electrophoresis. A. This figure shows how to seal between the gaskets and gel tray with agarose gel to prevent leaking. B. The separation difference between 0.7% and 1.4% agarose gel. While 0.7% agarose gel shows good separation on the TRF ladder, TRF ladder on 1.4% agarose gel is not separated well. 25 μl and 12.5 μl ladder was loaded in each lane of 0.7% and 1.4% gels, respectively. TRF ladder 1 and 2 in 1.4% gel are the same ladders. TRF ladder units are kilobase (kb).
- Add 5 μl loading dye to the samples. While 0.5x TBE can be used to analyze less than ~8 kb, 1x TAE buffer can be used for the ones that have longer telomeres (8 to 20 kb range) to have better separation.
- DNA Hybridization:
- Denature the gel for 20 min in 1.5 M NaCl and 0.5 M NaOH solution (pH 13.2) in a Pyrex® container (slowly shake). Make sure that denaturing solution covers the gel during shaking.
- Rinse gel with MilliQ® water to remove NaOH.
- Put the gel upside down on 2 sheets of 3MM Whatman® paper and wrap on the top of the gel (Figure 3A and B).

Figure 3. The method for drying the gel (A and B). Whatman® papers are located at the bottom, gel is located in between Whatman® papers and plastic wrap is on top of the gel. - Dry the gel using a gel dryer (56 °C for ~3 h).
- Transfer the gel to a Pyrex® container, rinse with MilliQ® water and remove the Whatman® paper.
- Neutralize the gel for 30 min with 0.5 M Tris-HCl and 1.5 M NaCl solution (pH 8). Make sure that neutralization solution covers the gel during shaking.
- Wrap gel around 25 ml pipette and transfer gel to a cylindrical hybridization tube.
Note: Make sure that caps are sealed well and don’t leak. - Prehybridize the gel with 10 ml hybridization solution for at least 10 min at 42 °C in hybridization oven.
- Add 12.5 μl hot probe (see step 7 at below) to a fresh 10 ml hybridization solution and let it hybridize overnight at 42 °C rotating hybridization oven.
Notes:- Be careful when you are working with radioactive material. Protect yourself with a shield and try to avoid any contamination to the work area.
- Remove hot hybridization buffer and keep it for next use (it can be used two times or place into radioactive liquid waste).
- Be careful when you are working with radioactive material. Protect yourself with a shield and try to avoid any contamination to the work area.
- Denature the gel for 20 min in 1.5 M NaCl and 0.5 M NaOH solution (pH 13.2) in a Pyrex® container (slowly shake). Make sure that denaturing solution covers the gel during shaking.
- Hot probe (C-rich) preparation
- Preannealed template
3.4 μl of 10 pmol/μl (10 μM) GTU4 oligonucleotide (GTU4 primer: 5’ -GGG UUA GGG UUA GGG UUA GGG AAA- 3’)
15.6 μl of 100 pmol/μl (100 μM) T3C3 + 9 oligonucleotide (T3C3 + 9 primer: 5’- TTT CCC TAA CCC TAA-3’)
1 μl of 1M NaCl (50 mM final concentration)
Cycler program:
Heat to 99 °C 1 min
37 °C 15 min
25 °C 15 min
Stored at -20 °C - 8x Adjusted Buffer M
500 μl 10x Buffer M
100 μl 2 M Tris.HCl (pH 7.4-7.6)
25 μl BSA (10 mg/ml) - Reaction
3.125 μl 8x Adjusted Buffer M
1.0 μl pre-annealed template oligo
2.5 μl dATP (0.5 mM)
2.5 μl dTTP (0.5 mM)
9.88 μl H2O (DEPC)
5.0 μl α-P32 dCTP
1.0 μl Klenow Exo:
25 °C for 30 min
98 °C for 5 min
25 °C for 5 min
Add 0.5 μl Uracil DNA glycosylase 1 U/μl (UDG)
37 °C for 10 min
95 °C for 10 min
Store the probe for no more than 2 weeks at -20 °C.
- Preannealed template
- Washing: Wash the gel once in 2x SSC, 0.1% SDS solution for 15 min at 42 °C, then wash the gel twice in 0.5x SSC, 0.1% SDS solution for 15 min at 42 °C. Finally, wash the gel twice in 0.5x SSC, 1% SDS for 15 min at 42 °C. 10-15 ml washing solution can be used for each washing step at 42 °C rotating hybridization oven.
Note: Prepare the washing solutions in the following order: SSC, water, SDS so they dissolve easily. - Exposure: Prepare the gel for scanning. Briefly, wrap the gel with plastic (Saran type) wrap in the cassette and put the screen on the gel (Figure 4). Expose it at least 4 h, preferably overnight. Scan the screen on Typhoon PhosphorImager.

Figure 4. Illustrates the preparation of the gel for scanning step by step. Prepare the saran plastic wrap in an appropriate cassette size and spread it into the cassette. Then, put gel onto this plastic wrap and cover the gel by wrapping with additional plastic wrap. Place screen on top of the gel that is surrounded plastic wrap, then incubate it in a dark place. All steps should be done behind a protective shield. - Calculate the TRF lengths from phosphor Imager scans.
Description of telomere length measurements by using Image Quant® software and Graph Pad Prism 6® together.- Measure the distance for each marker band from the top in Image Quant software (X: measured distance, Y: molecular weight) (Figure 5A).
- Draw a rectangle with 150 rows around the samples and background (empty lane) in Image Quant software (Figure 5B).
- Get the intensity of each 150 boxes for each sample (Volume reports from analysis tool give the intensity values) in Image Quant software. Export volume values from Image Quant to Excel.
- Open Graph Pad Prism and create a new data and table. Check “Y: Enter and plot a single Y value for each point”. First column (X): measured distance, second column (Y): molecular weight. Go to analysis, analyze, XY analyses, nonlinear regression (curve fit) and select one phase exponential decay. Go to tab “Range”: check “create a table of XY coordinates of 150 points that define the curve”. Nonlinear fit of Data1 contains molecular weight for each distance in the Y row.
- Create a new page with new data and table in Graph Pad Prism. Check “Y: Enter and plot a single Y value for each point”. Copy background volume values (intensity) from excel into first column and sample values into second column. Go to analysis, Analyze, double click “transform”. Check “Transform Y values using Y=Y-X”. This equality gives the transform of data 2 [new intensity (Y)= sample intensity (Y) – background intensity (X)]. Go to analysis, analyze, column analysis, double click “column statistics”, click ok. This gives sum: Σ(Inti).
- Create a new page with new data and table in Graph Pad Prism. Check “Y: Enter and plot a single Y value for each point”. Copy and paste nonlinear fit of Data 1 (Y) into X column. Copy and paste Transform of Data 2 (Y) into Y column. Go to analysis, Analyze, double click “transform”. Check “Transform Y values using Y=Y/X”. Go to analysis, analyze, column analysis, double click “column statistics”, click ok. This gives sum: Σ(Inti/MWi). MW: molecular weight.
- Calculate the average telomere length using following formula =Σ(Inti)/ Σ(Inti/MWi). Figure 5 C and D show an example for TRF gel and average telomeric lengths for some non-small cell lung cancer cell lines, respectively.
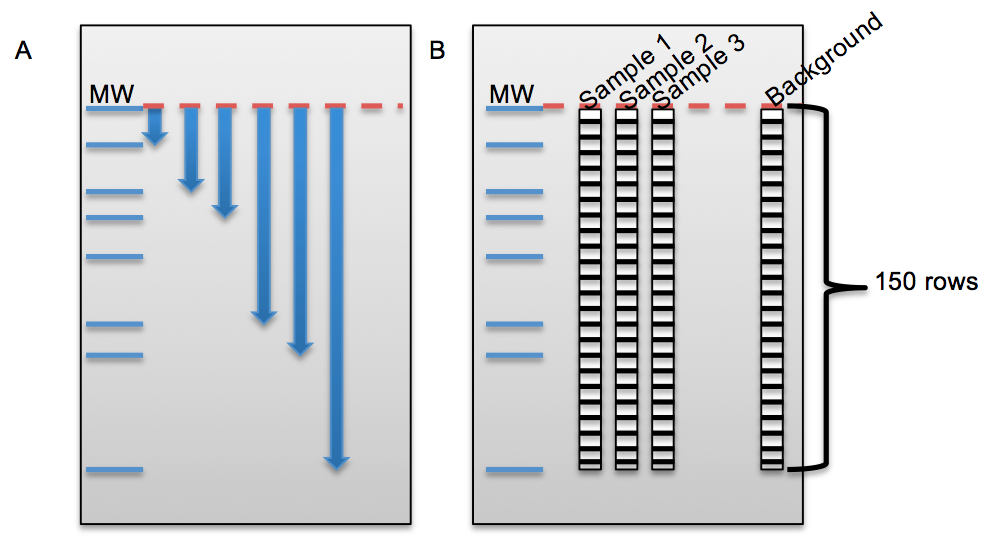
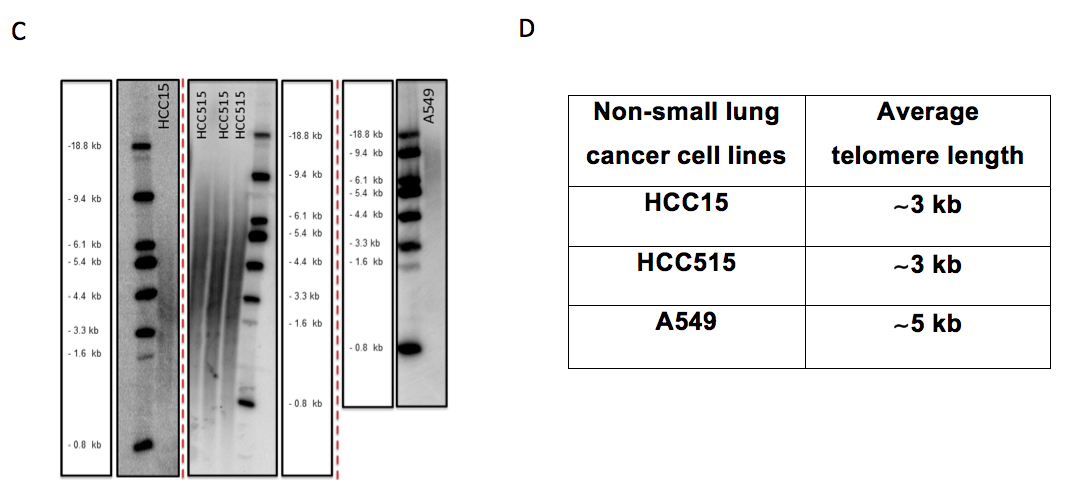
Figure 5. The beginning steps for telomeric length measurement in Image Quant and some examples for non-small cell lung cancer cell lines on different TRF gels. A. Figure shows the distance measurement for each molecular weight (MW) from top baseline (red dash line) in Image Quant. B. Figure shows columns with 150 rows on each sample and empty lane (background) to calculate the intensity (volume) of each lane in Image Quant. C. HCC15, HCC515 and A549 nonsmall cell lung cancer cell lines on different TRF gels with molecular weight (MW) marker. D. Average telomere length for HCC15, HCC515, A549. The unit for ladder sizes is kilobase (kb).
- Measure the distance for each marker band from the top in Image Quant software (X: measured distance, Y: molecular weight) (Figure 5A).
Recipes
- 10x PBS buffer-phosphate buffer saline
Powder for 5 L of 10x is ready to use for preparation of 5 L of concentrated 10x phosphate-buffered saline (PBS)
Prepare 5 L milliQ® water and add PBS powder
Next add large stir bar and place on stirrer until solids are dissolved - 1x TAE buffer-Tris-Acetate-EDTA
Prepare 2% 50x Tris-Acetate-EDTA in milliQ® water - 5x TBE buffer-Tris-Borate-EDTA
Prepare a 5x stock solution (pH 8.3) in 10 L of MilliQ® water
540 g of Tris Base
275 g of boric acid (use a mask)
200 ml of 0.5 M EDTA (pH 8.0)
Add large stir bar and place on stirrer until solids are dissolved - Hybridization solution
30% (v/v) 20x SSC
5% 100x Denhardt
2.5% SDS - 100x Denhardt solution
Ficoll 5 g
Polyvinylpyrrolidone 5 g
BSA 5 g
dH2O 250 ml - 6x Glycerol and Bromophenol blue Loading Dye (50 ml)
15 ml glycerol (30% final con.)
125 mg bromophenol blue
34 ml DEPC H2O
~1 ml Tris HCl (pH 7.6)
Acknowledgments
Some of these protocols were adapted from previously published studies (Herbert et al., 2003). We thank Zeliha Gunnur Dikmen for her help in acquisition of TRAP gel and Abhijit Bugde from the Live Cell Imaging Facility at UT Southwestern for his assistance with the imaging and analysis part of Telomere dysfunction Induced Foci (TIF) analysis.
References
- Harley, C. B., Futcher, A. B. and Greider, C. W. (1990). Telomeres shorten during ageing of human fibroblasts. Nature 345(6274): 458-460.
- Hayflick, L. and Moorhead, P. S. (1961). The serial cultivation of human diploid cell strains. Exp Cell Res 25: 585-621.
- Herbert, B. S., Shay, J. W. and Wright, W. E. (2003). Analysis of telomeres and telomerase. Curr Protoc Cell Biol Chapter 18: Unit 18 16.
- Mender, I. and Shay, J. W. (2015a). Telomere dysfunction induced foci (TIF) analysis. Bio-protocol 5(22): e1656.
- Mender, I. and Shay, J. W. (2015b). Telomerase repeated amplification protocol (TRAP). Bio-protocol 5(22): e1657.
- Olovnikov, A. M. (1973). A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J Theor Biol 41(1): 181-190.
- Ouellette, M. M., Liao, M., Herbert, B. S., Johnson, M., Holt, S. E., Liss, H. S., Shay, J. W. and Wright, W. E. (2000). Subsenescent telomere lengths in fibroblasts immortalized by limiting amounts of telomerase. J Biol Chem 275(14): 10072-10076.
- Shay, J. W. and Wright, W. E. (1996). Telomerase activity in human cancer. Curr Opin Oncol 8(1): 66-71.
- Shay, J. W. and Wright, W. E. (2001). Telomeres and telomerase: implications for cancer and aging. Radiat Res 155(1 Pt 2): 188-193.
- Shay, J. W., Wright, W. E. and Werbin, H. (1991). Defining the molecular mechanisms of human cell immortalization. Biochim Biophys Acta 1072(1): 1-7.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Mender, I. and Shay, J. W. (2015). Telomere Restriction Fragment (TRF) Analysis. Bio-protocol 5(22): e1658. DOI: 10.21769/BioProtoc.1658.
Category
Cancer Biology > Replicative immortality > Cell biology assays > Nucleic acid sequencing
Cancer Biology > Replicative immortality > Cell biology assays > Protein
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.