- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Two-photon Photoactivation to Measure Histone Exchange Dynamics in Plant Root Cells
Published: Vol 5, Iss 20, Oct 20, 2015 DOI: 10.21769/BioProtoc.1628 Views: 8583
Reviewed by: Tie LiuVinay PanwarAnonymous reviewer(s)
Original research article
The authors used this protocol in:

Dec 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
Chromatin-binding proteins play a crucial role in chromatin structure and gene expression. Direct binding of chromatin proteins both maintains and regulates transcriptional states. It is therefore important to study the binding properties of these proteins in vivo within the natural environment of the nucleus. Photobleaching, photoactivation and photoconversion (photoswitching) can provide a non-invasive experimental approach to study dynamic properties of living cells and organisms. We used photoactivation to determine exchange dynamics of histone H2B in plant stem cells of the root (Rosa et al., 2014). The stem cells of the root are located in the middle of the tissue, which made it impossible to carry out photoactivation of sufficiently small and well-defined sub-cellular regions with conventional laser illumination in the confocal microscope, mainly because scattering and refraction effects within the root tissue dispersed the focal spot and caused photoactivation of too large a region. We therefore used 2-photon activation, which has much better inherent resolution of the illuminated region. This is because the activation depends on simultaneous absorption of two or more photons, which in turns depends on the square (or higher power) of the intensity-a much sharper peak. In this protocol we will describe the experimental procedure to perform two-photon photoactivation experiments and the corresponding image analysis. This protocol can be used for nuclear proteins tagged with photoactivable GFP (PA-GFP) expressed in root tissues.
Keywords: Histone exchangeMaterials and Reagents
- Sterile 9 cm square Petri dishes for plant growth media
- Cover slips No 1.5 (VWR International, catalog number: 631-0125 )
- Microscope slides with frosted end (VWR International, Superfrost®, catalog number: 631-0114 )
- Secure Seal Adhesive Sheets (0.12 mm thick) (Grace Bio-Labs, Inc., catalog number: 620001 )
- Arabidopsis lines expressing a protein of interest tagged with PA-GFP (Patterson and Lippincott-Schwartz, 2002)
Note: In our case, for measuring histone exchange dynamics in the root stem cells we placed the H2B-PAGFP construct under the control of a root cell-specific promoter (pSCR) (Rosa et al., 2014), which derives expression simultaneously in quiescent centre cells, endodermis/cortex initials, and root endodermis. This setup allowed us to compare the dynamics of a histone protein in pluripotent stem cells as they progress into more differentiated states. In this protocol we will not mention particular cells or tissues as the procedure can be applied to any cell type in the root. - Murashige & Skoog Basal Medium with Vitamins (any equivalent source would be suitable) (PhytoTechnology Laboratories®, catalog number: M519 )
- 10% bleach (contains 5-10% sodium hypochlorite) in dH2O (Vortex, Procter & Gamble)
- NH4NO3 (pH 5.8) (ForMediumTM)
- Sucrose (Sigma-Aldrich)
- Phytagel (Sigma-Aldrich, catalog number: P8169 )
- Murashige and Skoog medium (see Recipes)
Equipment
- Plant growth chamber
- Two-photon photoactivation experiments were performed on an Ultima two-photon laser scanning microscope (Buker Coopration, Prairie Technologies, model: Ultima 2-Photon Microscope )
Note: Live images were acquired using Olympus 60x 0.9 NA water immersion objectives at 512 x 512 resolution (0.203 μm/pixel) and 1 μm focal steps. - Laminar flow cabinet
Software
- ImageJ software
- MS Excel
- Graphpad prism software
Procedure
- Plant growth and microscope slide preparation
- Surface sterilize seeds in 5% v/v sodium hypochlorite for 5 min and rinse three times in sterile distilled water.
- Plate the seeds on to Murashige and Skoog medium (pH 5.8) supplemented with 1% w/v sucrose and 0.5% w/v Phytagel.
- Stratify the seeds by incubating for 2 days at 4 °C in darkness and then grown at 25 °C in continuous light in vertically oriented Petri dishes for approximately 3-5 days.
- Slide preparation: Cut small square frames of Secure Seal Adhesive with similar dimensions to the cover slip and stick it to the slide (this will avoid squashing the roots between the slide and cover slip and prevents the evaporation of mounting media during imaging) (Figure 1).
- Cut root tips from Arabidopsis seedlings whilst still on plates and place onto the microscope slide containing a drop of water. Alternatively, if seedlings are not too big, the whole seedling can be mounted on the slide. We mounted typically 1-2 roots per slide, but used only one of them for the 1 h time course, as we tried to avoid having the material too long on the slide prior to the experiment.
- Add a coverslip and stick it to the slide with the adhesive tape.
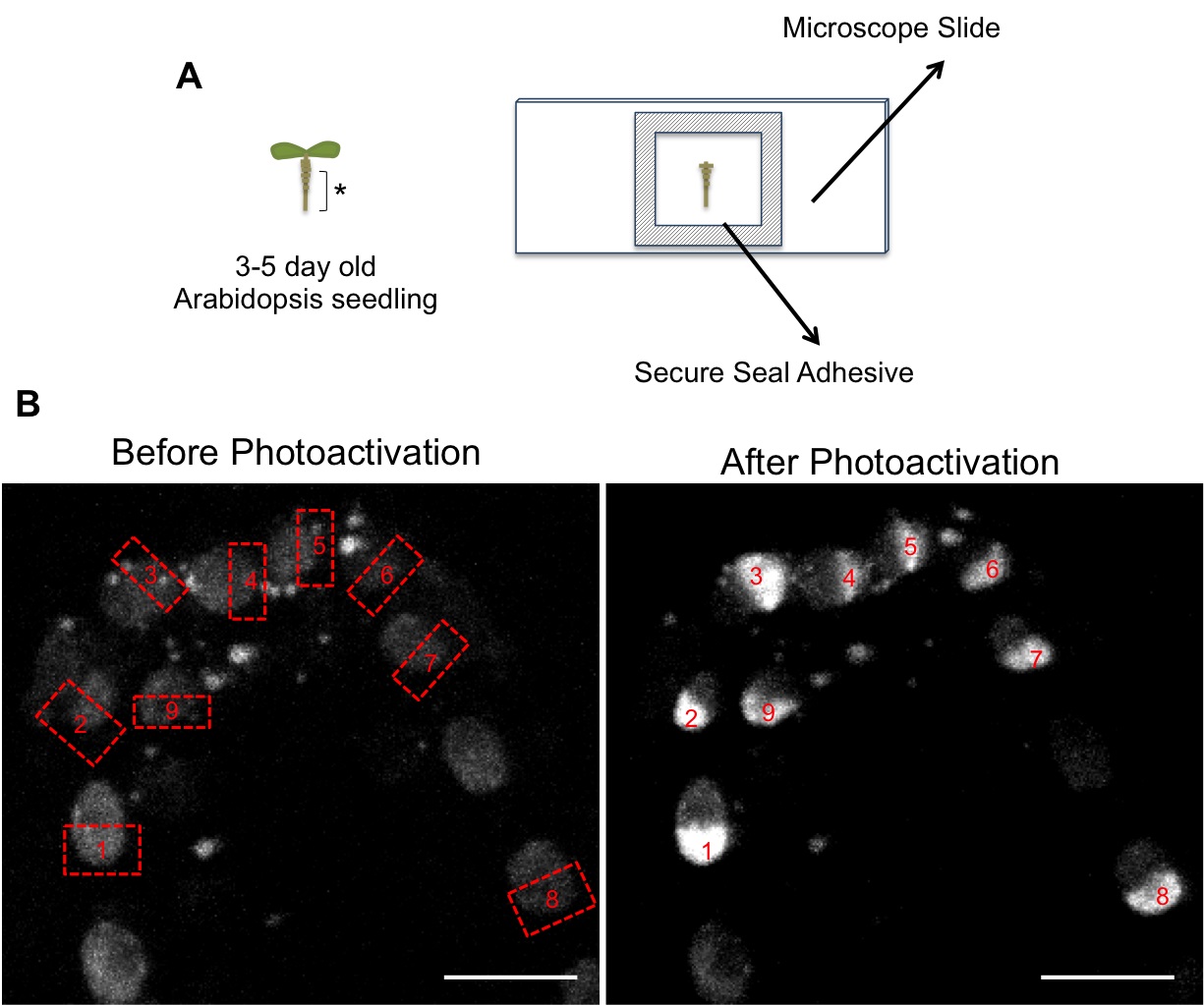
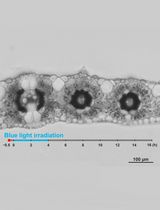
Figure 1. A. Schematic of microscope slide preparation. Arabidopsis root tips (asterisk) were cut and placed onto slides. B. Images showing selection of regions for photoactivation (labeled from 1 to 9) in roots expressing H2B-PAGFP. Before photoactivation nuclei show a very weak signal due to the faint pre-activation (see step B4). Scale bar = 5 μm.
- Surface sterilize seeds in 5% v/v sodium hypochlorite for 5 min and rinse three times in sterile distilled water.
- Imaging
- Two-photon photoactivation experiments were performed on an Ultima two-photon laserscanning microscope. Images were acquired using an Olympus 60x 0.9 NA water immersion objective.
- Use 2.5x optical zoom and 512 x 512 resolution.
- Set up Z-steps of 1 μm.
- The absence of fluorescence prior to photoactivation makes it is impossible to identify the regions of interest, therefore an initial pre-activation is necessary. This faint pre-activation is achieved by scanning the root tip at 850nm wavelength, 50% power and with a pixel time of 5.6 μs. One or two scans were necessary depending on the root (Figure 1B).
- For photoactivation define simultaneously several regions-of-interest (ROI) covering half of each nucleus in the different Z- positions. Pulse the ROIs with a 710 nm laser light at 30% power with a pixel time of 8.8 μs. Repeat this cycle two times, and then set up normal time series.
- For PA-GFP time series imaging use a 925 nm laser line at 50% power, 8.8 μs/pixel, with one acquisition (one z-stack, same z-step) every 60 sec for 1 h.
- Two-photon photoactivation experiments were performed on an Ultima two-photon laserscanning microscope. Images were acquired using an Olympus 60x 0.9 NA water immersion objective.
- Image processing
- Image analysis was performed using Image J software.
- Open the hyperstack (containing z and t dimensions) and apply a Z-stack maximum projection.
- It is expected that cell movements and/or drift will occur during image acquisition. These movements can be corrected and/or eliminated by aligning consecutive frames using StackReg plug-in of Image J. This plugin aligns successive image frames by a cross-correlation procedure, which works easily and without operator intervention, as long as movements are not too large. More details, including screen shots are available on the developers’ website. (http://bigwww.epfl.ch/thevenaz/stackreg/)
- Once images are aligned using the “ROI manager” tool from Image J to define three regions of interest: Photoactivation Region; Total Nucleus and Background (Figure 2A). Both the Photoactivation and Total Nucleus Regions are drawn by hand based on contrast of fluorescence. Because mean intensities are measured the exact outline of each area is not critical for the result.
- Once the three regions are loaded onto the “ROI manager” window click “Multi Measure” and a results table will be provided with the mean intensity values for each region at the different time points. These measurements will be used later for normalizing the data.
- Image analysis was performed using Image J software.
- Data analysis
- Export the raw intensity measurements to Microsoft Excel to perform a double normalization (Phair et al., 2004). This method normalizes the initial fluorescence to “1” and corrects for bleaching during imaging (Figure 2).
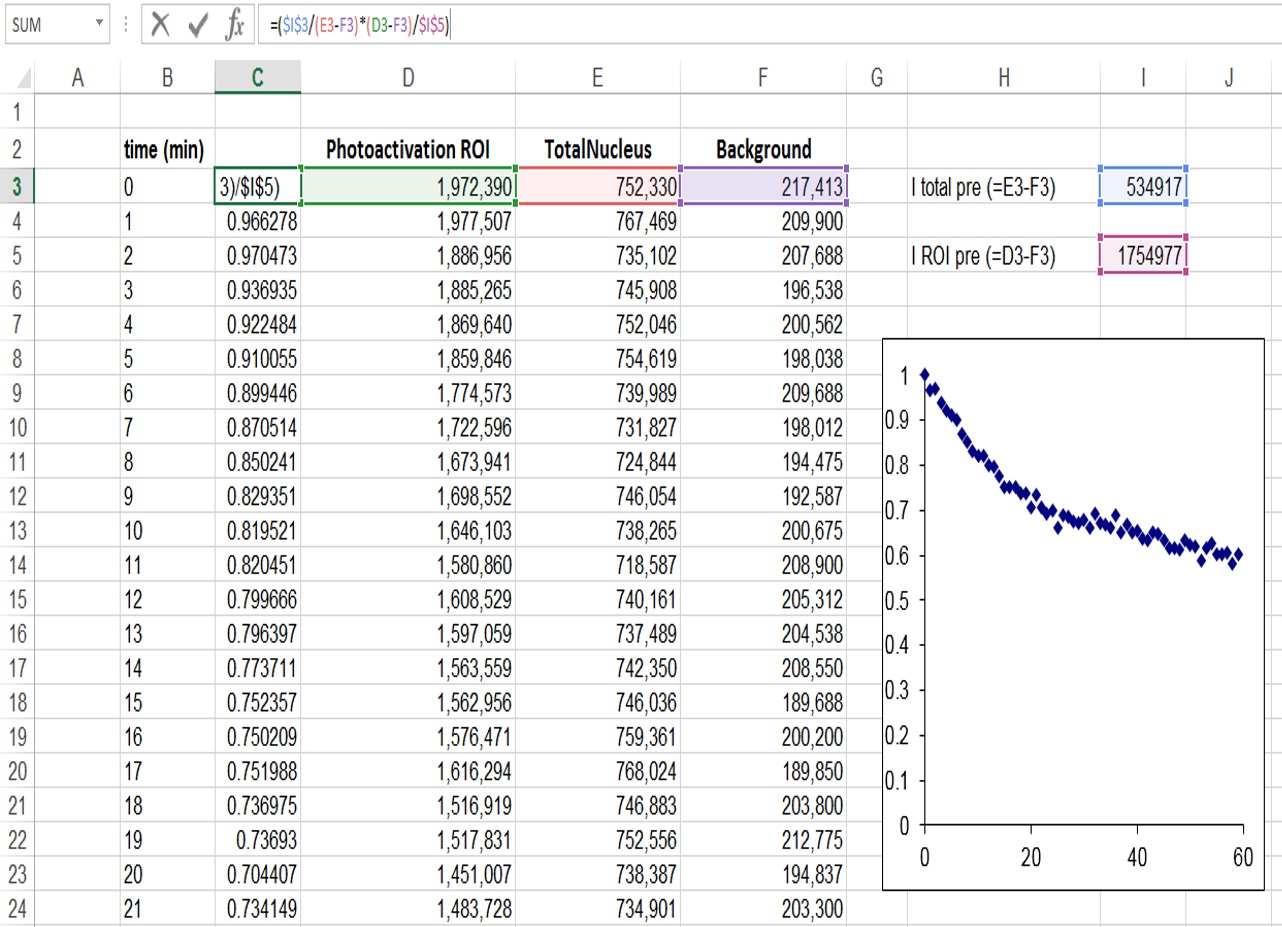
Figure 2. Determination of relative fluorescence signal in the ROI region, and correction for background and bleaching during imaging - It is also important to normalize for the proportion of the nucleus that is activated (Figure 3). In our case because half of the nucleus was activated the fluorescence decay could only reach at most half of the initial fluorescence. We therefore took this into account by additionally normalizing as follows: I (norm) = (Ft-F0/2)/(F0/2), where Ft is the fluorescence intensity at each time point (t) and F0 is the initial fluorescence intensity after photoactivation.
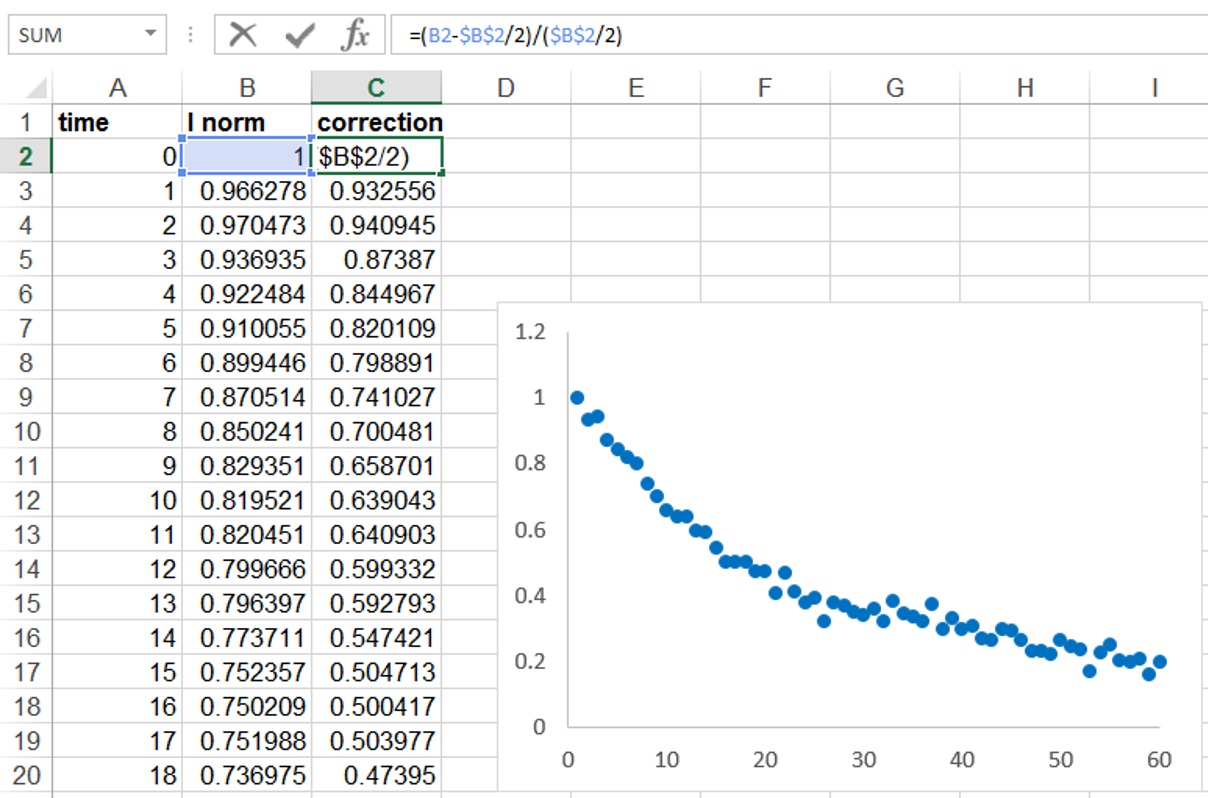
Figure 3. Correction for the proportion of the nucleus activated - Copy the normalized data to a program such as GraphPad Prism and perform a curve fitting analysis. Use a non-linear regression and to a single exponential decay function (Figure 2C). The mobile fraction (Mf) and half-life values (the time it takes for fluorescence intensity to reach half the maximum of the plateau level) are obtained by the software.
- Present data as the average ± SEM and assess the statistical significance by t-test.
Note: The examples regarding decays at different developmental stages can be found in our publication (Rosa et al., 2014).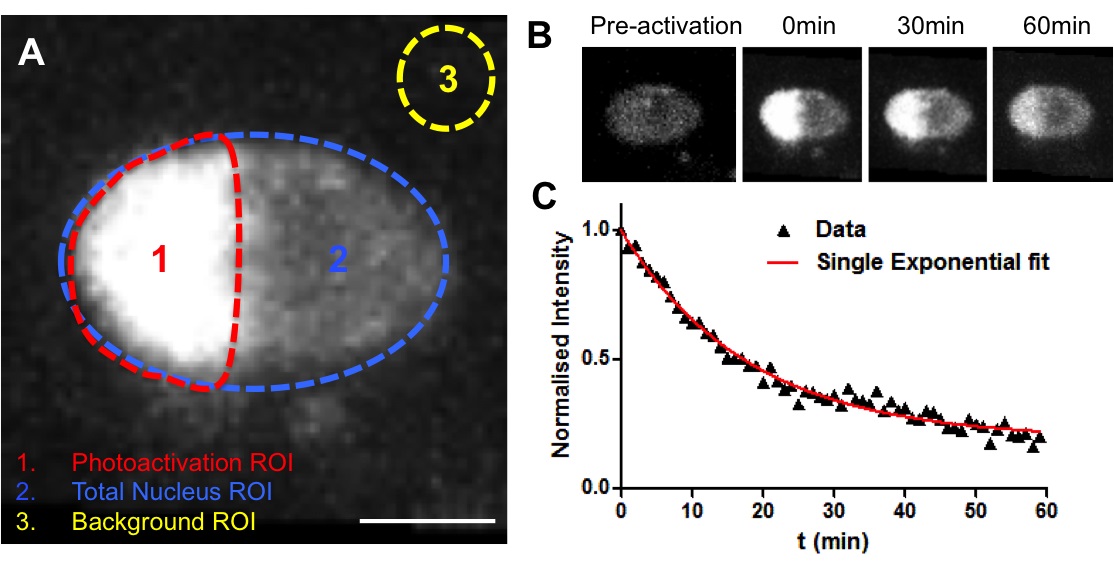
Figure 4. Example of a photoactivation experiment. A. Establishment of three ROIs for analysis (Photoactivation region, Total nucleus and background). B. Representative fluorescence changes in a nucleus from the endodermis expressing H2B-PAGFP. Half of the nucleus was photoactivated (710 nm laser line) and the loss of fluorescence within the activated area was measured. C. Decay curve for nucleus depicted in B. Data corresponded to fluorescent intensities at the photoactivated region (1) after normalisation. In red, non-linear regression using a single-exponential function.
- Export the raw intensity measurements to Microsoft Excel to perform a double normalization (Phair et al., 2004). This method normalizes the initial fluorescence to “1” and corrects for bleaching during imaging (Figure 2).
Recipes
- Murashige and Skoog medium
0.025 mg/L CoCl2.6H2O
0.025 mg/L CuSO4.5H2O
36.7 mg/L Na Fe-EDTA
6.2 mg/L H3BO3
0.83 mg/L KI
16.9 mg/L MnSO4.2H2O
0.25 mg/L Na2MoO4.2H2O
8.6 mg/L ZnSO4.7H2O
332.02 mg/L CaCl2.2H2O
170 mg/L KH2PO4
1,900 mg/L KNO3
180.5 mg/L MgSO4.7H2O
1,650 mg/L NH4NO3 (PH 5.8)
1% sucrose
0.5% phytagel
Acknowledgments
We thank Nuno Moreno for the help with the multiphoton microscope at Instituto Gulbenkian de Ciência, Portugal. This work was supported by grant (SFRH/BD/23202/2005) from the Portuguese Fundação para a Ciência e a Tecnologia, by the Biotechnology, and Biological Sciences Research Council of the UK (grant BB/D011892/1 and BB/J004588/1) and the John Innes Foundation.
References
- Patterson, G. H. and Lippincott-Schwartz, J. (2002). A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297(5588): 1873-1877.
- Phair, R. D., Gorski, S. A. and Misteli, T. (2004). Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy. Methods Enzymol 375: 393-414.
- Rosa, S., Ntoukakis, V., Ohmido, N., Pendle, A., Abranches, R. and Shaw, P. (2014). Cell differentiation and development in Arabidopsis are associated with changes in histone dynamics at the single-cell level. Plant Cell 26(12): 4821-4833.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Rosa, S. and Shaw, P. (2015). Two-photon Photoactivation to Measure Histone Exchange Dynamics in Plant Root Cells. Bio-protocol 5(20): e1628. DOI: 10.21769/BioProtoc.1628.
Category
Plant Science > Plant cell biology > Cell imaging
Cell Biology > Cell imaging > Two-photon microscopy
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.