- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Morphological Quantification of Nuclei and Mitochondria in Serial Block-face Scanning Electron Microscopy Images
Published: Vol 5, Iss 18, Sep 20, 2015 DOI: 10.21769/BioProtoc.1593 Views: 9532
Reviewed by: Anonymous reviewer(s)
Original research article
The authors used this protocol in:

Jul 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics








Abstract
Serial Block-face Scanning Electron Microscopy (SBF-SEM or 3D-EM) is a powerful tool to study biological structure in ultrastructural level. Quantification of cellular ultrastructure is useful to providing biological information. This technique requires not only high quality of tissue fixation and ideal sample embedding to preserve structures, but also delicate 3D image scanning and post-processing of images. We have adapted previous method to optimize the EM technique to detect and study cellular ultrastructure. Here we present the method to embed samples for 3D-EM technique and to quantify the morphological parameters of nucleus and mitochondria.
Part I. Tissue embedding for 3D-EM images
Materials and Reagents
- Adult mouse brain / spinal cord
- 4% Paraformaldehyde (PFA) (Sigma-Aldrich, catalog number: P6148 )
- Glutaraldehyde solution (GA) (Sigma-Aldrich, catalog number: G5882 )
- Sodium Cacodylate Buffer (0.4 M, pH 7.2) (Electronic Microscopy Science, catalog number: 11654 )
- Phosphate buffered saline (PBS) (1x Solution, Fisher BioReagents) (Fisher Scientific, catalog number: BP2438-4 )
- Potassium ferricyanide (III) (Sigma-Aldrich, catalog number: 702587 )
- Thiocarbohydrazide (TCH) (Electronic Microscopy Science, catalog number: 21900 )
- Osmium tetroxide 4% solution (Electronic Microscopy Science, catalog number: 19150 )
- Uranyl acetate (UA) (Electron Microscopy Sciences, catalog number: 22400 )
- Lead Nitrate (Crystalline/Certified ACS), Fisher Chemical (Fisher Scientific, catalog number: L62-100 )
- L-aspartic acid solution (Sigma-Aldrich, catalog number: A9256 )
- Ethanol, 200 proof (100%), USP, DeconTM Labs (Fisher Scientific, catalog number: 07-678-004 )
- Acetone (Electronic Microscopy Science, catalog number: 10010)
- EMbed-812 kit (Electronic Microscopy Sciences, catalog number: 14120 )
- EMS Molded Flat Embedding Mold (Electronic Microscopy Sciences, catalog number: 70905-01 )
Equipment
- Vibratome (Leica, catalog number: Leica VT1000S )
- Zeiss Sigma VP SEM (Zeiss) with Gatan 3View (Gatan)

Figure 1. Zeiss Sigma VP SEM (Zeiss) with Gatan 3View (Gatan)
Procedure
- Mouse is perfused intracardially with 20 ml PBS in 1-2 min followed by 50ml 4% PFA with 1% GA in 5 min.
- Mouse brain/spinal column is dissected out and post-fixed by 4% PFA with 1% GA for 24 h at 4 °C.
- Peel the spinal cord from spinal column.
- Fixed brain/spinal cord sections are made with 100 μm thickness on a vibratome.
- Brain/spinal cord sections are cut into 0.2 x 0.2 cm2 tissue blocks depending on the interesting area.
- Tissue blocks are put in 1.5 ml Eppendorf tubes and incubated in 1ml cold (4 °C) 0.1 M cacodylate buffer for 3 times for 5 min each on ice.
- Right before use, make fresh OsO4-ferrocyanide buffer by mixing 3% potassium ferricyanide in 0.2 M cacodylate buffer and an equal volume of 4% aqueous osmium tetroxide. The tissue blocks are incubated in this solution for 1 h on ice.
- During the one hour incubation, freshly prepare the fresh thiocarbohydrazide solution. Add 0.1 g thiocarbohydrazide (TCH) to 10 ml ddH2O and place in a 60 °C oven for 1 h. Agitate the buffer by swirling gently every 10 min to facilitate dissolving. Filter this solution through a 0.22 μm Millipore syringe filter right before use.
- At the end of the incubation in 0.2 ml OsO4-ferrocyanide, the tissues are incubated in ddH2O for 3 times for 5 min each at room temperature.
- Remove ddH2O in sample tubes and add TCH to sample tube directly after its incubation in 60 °C. Tissue blocks are incubated in TCH for 20 min at room temperature.
- Tissue blocks are incubated in 1ml ddH2O for 3 times for 5 min each, and then incubated in freshly prepared 0.2 ml 2% osmium tetroxide for 30 min at room temperature.
- Tissue blocks are incubated in 1 ml ddH2O for 3 times for 5 min each, and then incubated in 1% uranyl acetate overnight in 4 °C.
- The next day, prepare lead aspartate solution freshly. Firstly, prepare 0.03 M Aspartic Acid buffer. Briefly, add 0.2 g L-Aspartic Acid in 50 ml ddH2O. Adjust pH value to 5.5, and then put the Aspartic Acid solution at 60 °C until Aspartic Acid is totally dissolved. Secondly, prepare lead aspartate solution. Dissolve 0.066 g of lead nitrate in 10 ml aspartic acid solution. Put lead solution in a 60 °C oven for 30 min.
- Tissue blocks are incubated in 1ml ddH2O for 3 times for 5 min each, and then incubated in lead solution at 60 °C for 30 min.
- Tissue blocks are incubated in 1ml ddH2O for 3 times for 5 min each at room temperature, and then dehydrated using ice-cold solutions of freshly prepared 20%, 60%, 80%, 90%, 100% ethanol for 5 min each step.
- Tissue blocks are incubated in acetone for 10 min at room temperature.
- During acetone incubation, prepare 100% Epon solution using Epon-812 embedding kit as follows: mix EMbed-812 10 ml, Dodecenylsuccinic anhydride (DDSA) 8 ml, Methyl-5-Norbornene-2, 3-Dicarboxylic Anhydride (NMA) ml and 4 2, 2-dimethoxypropane (DMP-30) 0.4 ml together very well. To prepare 50% Epon solution, mix equal volumes of of 100% Epon with 100% acetone.
- Tissue blocks are incubated in 50% Epon solution for 2 h at room temperature.
- Tissue blocks are placed in 100% Epon solution on shaker overnight at room temperature.
- The next morning, prepare new 100% Epon solution using the recipe in 17. Transfer the tissues into new Epon solution, and then put them on shaker for 2 h at room temperature.
- Transfer Epon to Plastic Flat Embedding Mold and polymerize tissue blocks in a 60 °C oven overnight.
- Cut the samples put from Epon blocks and try to remove all the Epon surrounding the samples.
Note: It’s better to cut all the Epon away since Epon will cause charging during the scanning. The best size for the tissue block after the cutting is 1*1*2 mm3, but it’s very technique demanding and people seldom reach this standard.
- SBF-SEM images were acquired using a Zeiss Sigma VP SEM with Gatan 3 View. Serial image stacks of images at 100nm steps were obtained by sectioning (48 x 48 x 20) µm3 tissue blocks (length x width x depth) at a resolution of 8192 x 8192 pixels.
Note: Steps 1-20 should be done under a fume hood.
Representative data
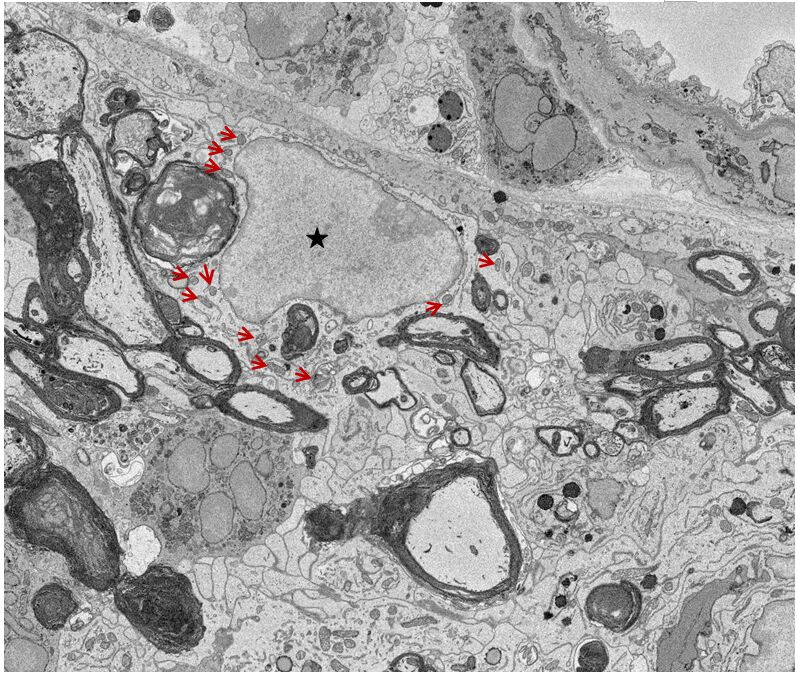
Figure 2. A representative single SBF-SEM image from a stack images showing nucleus and mitochondria in microglia. ↑: Mitochondria; ★: Nucleus.
Part II. Quantification of nuclei and mitochondria in 3D-EM images
Software
- Fiji software (NIH; http://fiji.sc/Downloads)
Procedure
- Save the images of the same stack in one folder.
- Open Fiji software and drag the folder to Fiji software to open the images.
- Click Plugins/Registration / Linear stack Alignment with SIFT to produce an aligned stack images.
- Save the stack images as tiff format.
- Click File/New/TrakEM2 (blank), and then choose the location to store the TrakEM file.
- Drag the Aligned stack images in tiff format to TrakEM image procession station.
- In Template window, right click anything / Add new child / area list.
- In Project Objects window, right click Untitled / Add / New anything.
- In project Objects window, right click anything / Add / New area_list.
- Right click New area_list / Rename to rename the layer as nucleus.
- In project Objects window, right click anything / Add / New area_list.
- Right click New area_list / Rename to rename the layer as mitochondria.
- In image processing station, right click the stack images, choose display / calibration, change the Unit of Length as micron, and put the Pixel Width, Pixel Height and Voxel Depth in the window. The Pixel Width and Pixel Height should be calculated according to the image scanning settings. The Voxel Depth is the interval of each image in the stack. For example, if the interval of the images is 100 nm, the Voxel Depth should be 0.1 micron.
- In Layers window, choose all the layers and right click the layers, choose Scale Z and thickness, calculate the scale number, which should put in the winder and could make the interval of each layer equal to the Voxel Depth.
- In image processing station, two layers named nucleus and mitochondria should appear in the left of the window.
- Click nucleus layer, double click the eyedropper tool to choose the one color.
- Use pen tool to draw the outline of nucleus in each image of the stack and click shift + F to fill the nucleus area in each image to make 3D reconstruction of the nucleus
- In control penal of Fiji, click Analyze / measure to measure the parameters of the nucleus including volume.
- Quantification of nuclear shapes is conducted in 3D reconstructions. Nuclei were categorized as follows; Round: round shape and smooth surface with ratio of length/width≤1.5; Elongated: elongated or oval shape with length/width>1.5, and may have small indentations; Bilobulated: two connected lobes with single intervening large indentation; Irregular: complicated shape with corrugated surface, and may have multiple and variable sizable indentations.
- Click mitochondria layer, double click the eyedropper tool to choose one color other than nucleus.
- Use pen tool to draw the outline of the mitochondria in each image of the stack and click shift + F to fill the outline are in each image to make 3D reconstruction of the mitochondria
- In control panel of Fiji, click Analyze / measure to measure the parameters of the mitochondria including volume and max diameter (Length of the mitochondria).
- Go through all the stack images, choose five different images where the cross section area of the mitochondria could be representative of the median of the cross section area.
- In the five representative images, use line tool and Analyze / measure in the control panel to draw and measure length of an Axis and b Axis.
- Use Cross section area = πab. Save all the parameters in a excel file. Calculate the mean Cross section area by the formula: Sum of five Cross section areas / 5.
- Repeat steps 11-12 to add another layer for the second mitochondria.
- Repeat steps 20-25 to do the quantification of mitochondria.
- Repeat steps 26-27 until finish the quantification of all the mitochondria in the cell.
- Calculate the number of mitochondria and the total volume of mitochondria in the cell.
Acknowledgments
The authors would like to thank the members of the Dr. Richard M. Ransohoff’s Lab for help on this manuscript. This work was supported by the National Multiple Sclerosis Society.
References
- TrackEM2 User Manual: http://www.ini.uzh.ch/~acardona/trakem2_manual.html
- TrakEM2 tutorials: http://fiji.sc/TrakEM2_tutorials
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Lu, H., Ohno, N. and Ransohoff, R. M. (2015). Morphological Quantification of Nuclei and Mitochondria in Serial Block-face Scanning Electron Microscopy Images. Bio-protocol 5(18): e1593. DOI: 10.21769/BioProtoc.1593.
Category
Cell Biology > Cell imaging > Fixed-tissue imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.