- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Genome-Wide siRNA Screen for Anti-Cancer Drug Resistance in Adherent Cell Lines
Published: Vol 5, Iss 10, May 20, 2015 DOI: 10.21769/BioProtoc.1474 Views: 13547
Reviewed by: HongLok LungMichael EnosAgnieszka Pastula
Original research article
The authors used this protocol in:

May 2014

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
The expression of genes is frequently manipulated in cell lines to study their cellular functions. The use of exogenous small Interfering RNAs (siRNAs) is a very efficient technique to temporarily downregulate the expression of genes of interest [reviewed by Hannon and Rossi (2004)]. A genome-wide siRNA library allows the user to study both the effect of each individual gene on a particular cell phenotype in a high throughput manner and also assess its phenotypic effect relative to all other genes targeted. Several factors that potentially influence the outcome of a screen need to be considered when performing a large siRNA screen (Jiang et al., 2011). Here we present a detailed protocol for a genome-wide screen to identify genes involved in anti-cancer drug resistance using the human siGENOME library from Dharmacon. In this protocol, we focus on resistance to treatment with the Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitor (EGFR-TKI) erlotinib in the lung cancer cell line PC9, which is exquisitely sensitive to EGFR-TKIs (de Bruin et al., 2014). This protocol can be used for other cell lines and other drug treatments, as we expand in the Notes below.
Materials and Reagents
- Human lung adenocarcinoma cell line PC9 (RIKEN BioResource Center, catalog number: RCB4455 )
- RPMI 1640 Medium containing L-Glutamine (Life Technologies, Gibco®, catalog number: A10491-01 )
Note: For cell culture, the medium is supplemented with 10% FBS and 1% Penicillin Streptomycin (see details FBS and Pen/Strep below). - Penicillin streptomycin (Life Technologies, Gibco®, catalog number: 15070-063 )
- Fetal bovine serum (FBS) (PAA Laboratories GmbH, catalog number: A15-101 )
- Opti-MEM® I Reduced Serum Medium (Life Technologies, Gibco®, catalog number: 31985-047 )
- 384-well tissue culture plates, black with clear F-bottom (4titude, Anachem, catalog number: 4TI-0201 were used for our screen; currently plates from Greiner bio-one (catalog number: 781091 ) are used
- Adhesive foil plate seal (Brandle Plate Sealer, Alpha Biotech)
- 5x siRNA buffer (GE Healthcare, catalog number: B-002000-UB-100 )
- Human siGENOME SMARTpool siRNA library (GE Healthcare, catalog number: G-005005 )
Note: We used an older version (from 2005) for our study (de Bruin et al., 2014) that is no longer available. Updated versions will contain other siRNA sequences as well as annotations, which will impact results. We therefore strongly recommend validating identified genes using other approaches. - siRNA controls (see Note 1)
Negative controls:Human siGENOME non-targeting siRNA 2 (GE Healthcare, catalog number: D-001210-02 ), RISC-Free control siRNA (catalog number: D-001220-01 ), ON-Targetplus non-targeting (catalog number: D-001810-10 )
Positive controls: Human siGENOME SMARTpool UBB (GE Healthcare, catalog number: M-013382-01 ) and PLK1 (Dharmacon, catalog number: M-003290-01 ) - DharmaFECT 2 siRNA transfection reagent (GE Healthcare, catalog number: T-2002-01 ) (see Note 2)
- DAPI (Roche Diagnostics, catalog number: 10236276001 )
- siGENOME LaminA/C control siRNA (GE Healthcare, catalog number: D-001050-01-05 ) (see Note 3)
- Human LaminA/C antibody (catalog number: SC-7292 ) (see Note 3)
- Phosphate buffered saline (PBS) (pH 7.0, made in-house)
- Erlotinib (Enzo Life Sciences, catalog number: BML-DL279 )
Equipment
- Acumen (TTP LABTECH, mode: eX3 )
- ArrayscanVTi HCS microscope (Cellomics)
- Biomek FX Liquid handling platform (Beckman Coulter)
- Countess Cell counter (Life Technologies)
- Liquid dispensor8-channel [for our screen the WellMate (Matrix) was used, currently the FluidX (Xrd-384) is used]
- Cytomat 24 °C CO2 incubator 37 °C and 5% CO2
Note: The Cytomat is a cell culture incubator that has racks built in holding ~500 plates, and the racks rotate to minimize plate edge effects. - Plate washer (BioTek Instruments, model: ELx405 Select CW , Figure 1)
- Plate sealer (Brandel Plate Sealer, Alpha Biotech)

Figure 1. Picture of plate washer
Important considerations
Before performing a large-scale siRNA screen, there are a few crucial points to consider and to optimize in order to obtain the best results (Jiang et al., 2011). These include:
- Optimization of the siRNA transfection: Reagent, cell number, siRNA concentration.
- Optimization of the drug dose, treatment time length in combination with the siRNA transfection for drug resistant/sensitizing screens.
Details of these optimization steps are provided in the Notes section at the end of this protocol. We recommend performing, if possible, a pilot screen with a range of random siRNAs and selected control RNAs to determine the feasibility and screen read-out prior to the genome-wide screen.
Procedure
- siRNA reverse transfection
- Culture PC-9 cells in RPMI medium, supplemented with 10% FBS + 1% Penicillin and Streptomycin.
- Resuspend the siRNA SMARTpools library in 1x siRNA buffer (dilute the 5x siRNA buffer using RNase/DNase-free water) to reach a final concentration of 375 nM.
Note: Upon arrival, the library was resuspended using 1x siRNA buffer into 3.75 µM stock solutions and stored at -20 °C for long-term storage. At this step in the protocol, the stock library was further diluted into 375 nM. - Aliquot the siRNA SMARTpools from library assay plates using the BiomekFX into 384-well assay plates at a final concentration of 11 nM/well (load 2.5 µl/well). Prepare all plates in triplicate for both the untreated and the drug-treated conditions. Use the first 4 columns of each plate for positive and negative siRNA controls (Figure 2 and Note 2), which can be aliquoted at the same time of the library or on the day of the transfection.
Note: The plates can be prepared beforehand, sealed and stored at -20 °C, making this a suitable stopping point. - On the day of the reverse transfection, thaw the plates at RT for 20 min, and spin briefly for 1 min at 1,000 rpm (180 x g) before removing the seal. Add the siRNA controls in the first 4 columns if not done so at step A3.
- Dilute DharmaFECT2 in Opti-MEM media (see Notes 2-4; we used 0.075 µl DharmaFECT2/3 µl) and add 3 µl per well using the 16-channel Xrd-384 dispenser at high speed. Tap the plates to ensure the solutions mix well, and incubate the plates at RT for 15 min.
- In the meantime, trypsinize and count the PC9 cells in quadruplicate using the Countess cell counter, and dilute the cell suspension appropriately. For PC9 cells, the final cell number was 500 cells per well in 80 µl. We therefore prepared 15 L RPMI (with FCS and Pen/Strep) containing 93.75E06 cells (see Note 5) for a total of 402 384-well plates, which are 6 replicates of the genome siRNA library (201 plates for the untreated condition and 201 plates for the drug-treated condition).
- Add 80 µl cell suspension per well using the 16-channel Xrd-384 dispenser at high speed.
Note: For an optimal reverse transfection, cells should be added to the siRNA mix at least 15 min and no more than 45 min after step A4. - Incubate cells in incubator at 37 °C for 48 h in a Cytomat incubator to ensure efficient protein knockdown.

Figure 2. Example of a layout of a 384-well plate. The first four columns were used for control siRNAs, with each row containing the siRNA indicated on the left. The first two columns were left untreated in the drug-treated conditions to confirm the effect of the drug. The remaining twenty columns were used for the genome-wide library, each well containing a different siRNA.
- Culture PC-9 cells in RPMI medium, supplemented with 10% FBS + 1% Penicillin and Streptomycin.
- Drug treatment
- Remove the medium using the Biotek PlateWasher (Figure 1); ensure that viable cells remain attached to the bottom of the wells.
- Add 80 µl RMPI (supplemented with FBS and Pen/Strep) per well with or without drug (see Note 6) using Xrd-384 dispenser at medium speed.
Note: For the treated condition, we left the first two columns containing control siRNAs without drug to determine the killing efficiency of the drugs in the experiment (Figure 2). - Incubate cells for a further 96 h (see Note 6).
- Remove the medium using the Biotek PlateWasher (Figure 1); ensure that viable cells remain attached to the bottom of the wells.
- Cell number quantification
- Remove media.
- Fix the cells by adding 90 µl of 80% ice-cold EtOH using Xrd-384 dispenser at low speed. Store plates for at least 1 h at -20°C.
Note: Overnight storage at -20 °C is perfectly fine and provides a suitable stopping point in the protocol. - Perform steps C15-18 in batches of 50 plates.
- Wash the plates 3x with 100 µl PBS using plate washer.
- Add 20 µl of a 1 µg/ml DAPI solution to each well and incubate the cells at RT for 1 h.
- Wash the plates with PBS using the plate washer.
- Seal the plates with adhesive foil seals.
Note: Plates can be stored at 4 °C prior to quantification. - Scan the plates using Acumen with the 405 nm laser to quantify the number of cells in each plate (see Note 7 and Figure 3).
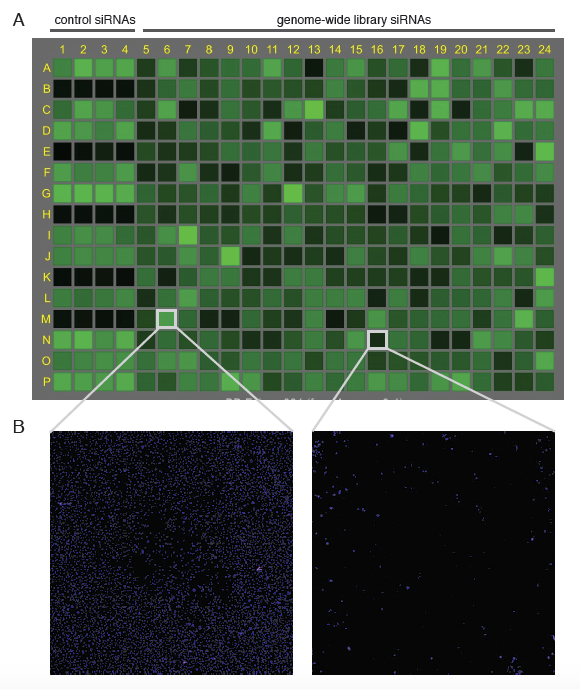
Figure 3. Example of Acumen output of a plate in untreated conditions. A. Once a plate has been scanned by Acumen using laser 405 nm, the number of stained cells is indicated in shades of green for each well. Note that the first four columns contain control siRNAs (see Figure 2). B. Each well can be visualized individually within the Acumen software package. Here, two examples are presented; the left well representing a well comparable to positive control siRNAs, which indicates that the siRNA did not impact, cell viability (>5,000 cells), whereas the right well shows the effect of an siRNA that impacted on cell viability (<500 cells).
- Remove media.
- Data Analysis
- Normalize the data within each plate using a robust Z-score calculation (Birmingham et al., 2009). For each plate, determine the median value of all samples (exclude the first four rows containing controls iRNAs) and subtract this value from each individual well. Divide each well by the median absolute deviation (MAD) of the plate to obtain a robust Z-score for each well. For example, the median sample value within plate A was 1,000 cells with an MAD of 200. If well D10 contains 800 cells, the robust Z-score for well D10 in plate A would be (800-1,000)/200=-1, which indicates that this well contains 1 deviation fewer cells than the median.
Normalize the data across the plates, separately for the untreated and drug-treated conditions. Determine for each position a smoothed Z-score using the median and MAD at each well position across all plates, in a similar manner as in the previous step. As mentioned above, calculate the smoothed Z-scores separately for the untreated and the drug-treated conditions. - Plot the smoothed Z-scores from the drug-treated condition against those from the untreated condition for each replicate (two conditions in triplicate gives nine comparisons in total).
- Determine the ‘line of best fit’ using linear regression.
- Calculate the residual difference between each data point (drug treated versus untreated) as the perpendicular distance between each data point and the ‘line of best fit’. A residual difference <0 indicate siRNAs were the viability within the drug condition is lower than would be expected based on the untreated condition, whereas a residual difference >0 indicate siRNAs were the viability within the drug condition is higher than would be expected based on the untreated viability. Thus, siRNAs with a positive residual difference indicate genes that reduce drug sensitivity upon knockdown of the gene.
Note: The results our genome-wide siRNA screen using this protocol are included in the original manuscript as Supplementary Tables (de Bruin et al., 2014). - For further validation, we selected siRNAs with a smoothed Z-score ≥ -2 in the untreated condition (no cell killing in the absence of the drug) and with a median residual difference ≥ 2 (desensitizing) or ≤ -2 (sensitizing). As described (de Bruin et al, 2014), this selection of siRNAs was further validated by performing a deconvoluted siRNA screen using the Dharmacon Set of 4 Upgrade siGENOME siRNAs.
- Normalize the data within each plate using a robust Z-score calculation (Birmingham et al., 2009). For each plate, determine the median value of all samples (exclude the first four rows containing controls iRNAs) and subtract this value from each individual well. Divide each well by the median absolute deviation (MAD) of the plate to obtain a robust Z-score for each well. For example, the median sample value within plate A was 1,000 cells with an MAD of 200. If well D10 contains 800 cells, the robust Z-score for well D10 in plate A would be (800-1,000)/200=-1, which indicates that this well contains 1 deviation fewer cells than the median.
Notes
- Positive and negative controls need to be determined for each cell line. For PC9 cells, we used siGENOME siRNAs targeting UBB (ubiquitin B) and siRNAs targeting PLK1 (polo-like kinase 1) as positive killing controls; siGENOME non-targeting siRNA2 (SC2), RISC-Free controlsiRNA (RF) and ON-Targetplusnon-targeting siRNA (ON-NT) were used as negative controls. In addition, we left two columns of control siRNAs untreated in the drug-treated plates and used these columns as treatment control (Figure 2).
- Optimal transfection reagent should be determined for each cell line, and depending on forward or reverse transfection. We tested 23 different transfection reagents in a reverse transfection in a 96-well format, using 0.1 and 0.3 µl for each reagent. We used LaminA/C siRNA and quantified both cell number and LaminA/C intensity and the fraction of cells exhibiting LaminA/C staining below a set threshold after staining with an LaminA antibody to assess toxicity and knock-down efficiency for each reagent as described in Note 3.
- Lamin A/C staining protocol using 96-well plates: Transfect cells as decribed above in steps A1-7, continue with steps C12-14. Then add 30 µl LaminA/C antibody 1:1,000 dilution in 3% BSA with PBS and incubate plates at RT for 1-2 h. Wash plates 3x with PBS and incubate plates with 30 µl Alexa Fluor488 donkey anti-mouse (1:1,500 dilution in 3% BSA with PBS). Wash plates 3x with PBS and follow with DAPI staining as described in steps C14-17. Scan DAPI (cell number) and LaminA/C intensity (knock-down efficiency) using the Acumen eX3 and Arrayscan VTi microscope.
- Having identified the transfection reagent (Note 2), the optimal concentration needs to be determined for use in 384-well plates. For PC9 cells, we tested 0.025, 0.035, 0.045, 0.055, 0.065 and 0.075 µl DharmaFECT 2 per well in 384 well format.
- Optimal cell seeding density should be determined for each cell line. For PC9 cells, we tested 500, 750, 1,000 and 1,250 cells per well in a 384-well plate. We aimed at 80-90% confluence at the end of the experiment in untreated condition.
- Optimal concentration and incubation time should be determined for each drug and each cell line. For erlotinib, we used 30 nM, a concentration slightly higher than the experimentally determined IC50 value for PC9 cells to bias towards detection of siRNA resulting in resistance.
- Preferentially, individual cells should be distinguishable in order to allow scanning by Acumen. The cell line should grow as single-cell layer and not form condensed clumps. Each cell line should be tested prior to performing a screen to determine if cell number can be quantified successfully by the Acumen or Arrayscan. We used the 405 nm laser with 6 mW output, 500 V PMT and sensitivity at 2. These settings depend on the lifetime of the laser and should be optimized for each cell line.
Acknowledgments
This protocol was developed for a study aiming to identify novel mechanisms of erlotinib resistance (de Bruin et al., 2014). E. B was funded by a fellowship from the Dutch Cancer Society and the High Throughput Screening facility at the Cancer Research UK-London Research Institute is funded by Cancer Research UK.
References
- Birmingham, A., Selfors, L. M., Forster, T., Wrobel, D., Kennedy, C. J., Shanks, E., Santoyo-Lopez, J., Dunican, D. J., Long, A., Kelleher, D., Smith, Q., Beijersbergen, R. L., Ghazal, P. and Shamu, C. E. (2009). Statistical methods for analysis of high-throughput RNA interference screens. Nat Methods 6(8): 569-575.
- de Bruin, E. C., Cowell, C., Warne, P. H., Jiang, M., Saunders, R. E., Melnick, M. A., Gettinger, S., Walther, Z., Wurtz, A., Heynen, G. J., Heideman, D. A., Gomez-Roman, J., Garcia-Castano, A., Gong, Y., Ladanyi, M., Varmus, H., Bernards, R., Smit, E. F., Politi, K. and Downward, J. (2014). Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov 4(5): 606-619.
- Hannon, G. J. and Rossi, J. J. (2004). Unlocking the potential of the human genome with RNA interference. Nature 431(7006): 371-378.
- Jiang, M., Instrell, R., Saunders, B., Berven, H. and Howell, M. (2011). Tales from an academic RNAi screening facility; FAQs. Brief Funct Genomics 10(4): 227-237.
Article Information
Copyright
© 2015 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
de Bruin, E. C., Jiang, M., Howell, M. and Downward, J. (2015). Genome-Wide siRNA Screen for Anti-Cancer Drug Resistance in Adherent Cell Lines. Bio-protocol 5(10): e1474. DOI: 10.21769/BioProtoc.1474.
Category
Cancer Biology > General technique > Cancer therapy > Genome-wide analysis
Molecular Biology > RNA > RNA interference
Molecular Biology > DNA > Gene expression
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.