- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

CytoTrap Two-Hybrid Screening Assay

Published: Vol 4, Iss 23, Dec 5, 2014 DOI: 10.21769/BioProtoc.1310 Views: 13813
Reviewed by: Tie LiuSaminathan Thangasamy
Original research article
The authors used this protocol in:

Apr 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
CytoTrap two-hybrid system provides an alternate strategy to detect protein-protein interactions in yeast. In this system, bait protein is fused with human son of sevenless (hSos) protein (Li et al., 1993), and a cDNA library or prey protein is expressed by fusion with myristoylation signal which anchors the prey fusion protein to yeast cell membrane. Protein interaction between bait and prey proteins recruits the hSos protein to the cell membrane, where hSos activates the Ras signaling pathway, leading to the survival of temperature-sensitive Saccharomyces cerevisiae (S. cerevisiae) strain cdc25H at 36 °C. In the CytoTrap two-hybrid system, detection of protein interaction occurs in the cytoplasm near cell membrane and is not dependent on transcription activation of reporter genes. Hence, the system is particularly useful for identifying interaction partners of transcription factors and proteins that need post-translational modification in the cytoplasm, which could not be used as bait proteins in conventional transactivation-based yeast two-hybrid systems. Here we describe the construction of a cDNA library from the model plant Arabidopsis and a procedure for screening interaction proteins of AtSR1/CAMTA3, a Ca2+/CaM-regulated transcription factor from this library. This procedure could be adapted to identify interacting partners of interested proteins from other organisms.
Keywords: Cytotrap systemMaterials and Reagents
- XL10-Gold Kanr Ultracompetent cells (Agilent, catalog number: 200317 )
- RNeasy plant mini kit (QIAGEN, catalog number: 74904 )
- Oligotex mRNA mini kit (QIAGEN, catalog number: 70022 )
- CytoTrap two-hybrid system (Agilent, catalog number: 200444 )
- YeastmakerTM yeast transformation system 2 (Takara Bio Company, Clontech, catalog number: 630439 )
- AccuScript (Agilent, catalog number: 200820 )
- RNase H (New England Biolabs, catalog number: M0297S )
- DNA polymerase I (New England Biolabs, catalog number: M0209S )
- UltraPureTM phenol: chloroform: isoamyl alcohol (25:24:1, v/v) (Life Technologies, catalog number: 15593-031 )
- T4 ligase (New England Biolabs, catalog number: M0202S )
- T4 polynucleotide kinase (New England Biolabs, catalog number: M0201S )
- Xho I (New England Biolabs, catalog number: R0146M )
- Acid-washed glass beads (Sigma-Aldrich, catalog number: G8772 )
- TritonTM X-100 (Thermo Fisher Scientific, catalog number: BP151-100 )
- Sodium dodecyl sulfate (SDS) (Thermo Fisher Scientific, catalog number: BP166-100 )
- Sodium chloride (NaCl) (J.T.Baker®, catalog nuber: JT3628-1 )
- Tris (J.T.Baker®, catalog number: JT4109-1 )
- EDTA (J.T.Baker®, catalog number: JT8993-1 )
- Yeast extract (Thermo Fisher Scientific, catalog number: BP9727-500 )
- Peptone (Thermo Fisher Scientific, catalog number: BP9725-500 )
- Dextrose (J.T.Baker®, catalog number: JT1919-1 )
- Adenine sulfate (Sigma-Aldrich, catalog number: A2545 )
- Yeast nitrogen base without amino acids (BD, catalog number: 291940 )
- DO supplement -His/-Leu/-Trp/-Ura (Takara Bio Company, Clontech, catalog number: 630425 )
- Histidine (Sigma-Aldrich, catalog number: H6034 )
- Tryptophan (Sigma-Aldrich, catalog number: T0254 )
- Galactose (Sigma-Aldrich, catalog number: G0750 )
- Raffinose (Sigma-Aldrich, catalog number: R0250 )
- Yeast lysis solution (see Recipes)
- YPDA/YPAD (see Recipes)
- SC/-LU (glucose) (see Recipes)
- SC/-LU (galactose) (see Recipes)
- 10x STE buffer (see Recipes)
Equipment
- Replica plating mold (Sunrise Science Products, catalog number: 3005-002 )
- RNase-free microcentrifuge tube (BioExpress, catalog numer: C-3262-2 )
- Microcentrifuge (Eppendorf, catalog number: 5424 )
- Shaker (New Brunswick Scientific, catalog number: M1299-0092 )
- Incubator (VWR International, catalog number: 414005-128 )
- 150-mm plate (VWR International, catalog number: 25384-326 )
- PCR thermocycler (Eppendorf, catalog number: 6321 000.515 )
Procedure
- Constructing and testing bait-hSos fusion protein
- DNA encoding bait protein is cloned into the pSos vector (plasmid map: http://www.chem-agilent.com/pdf/strata/217438.pdf) by the conventional enzyme-ligation method. The plasmid construct is confirmed by DNA sequencing to ensure the correct reading frame between hSos and bait.
- hSos-bait should be tested for auto-activation or interact with the myristylation signal prior to the CytoTrap two-hybrid screen (Aronheim et al., 1997).
- The pSos bait plasmid needs to be co-transformed into yeast host cell cdc25H with negative control either pMyr or pMyr-Lamin C using YeastmakerTM Yeast Transformation System 2.
- All the yeast culture procedures need to be conducted at room temperature (22-25 °C). After co-transformation, yeast cells are spread on synthetic media plate without leucine and uracil (SC/-LU) containing glucose, and incubate at room temperature for 4-5 days to allow yeast colonies to grow.
- Six independent colonies are picked and placed on SC/-LU medium plate containing galactose, and the plates are kept at room temperature for 1 h, and then transferred to 36 °C for 3 to 6 days for yeast growth.
- If the yeast cells co-transformed with the tested bait plasmid and empty pMyr or pMyr-Lamin C are able to grow at 36 °C, then it will be considered unsuitable for the CytoTrap two-hybrid system.
- Only bait plasmids that do not lead to growth of host yeast cells at 36 °C in the co-transformation test could be suitable to screen for interaction partners using the CytoTrap two-hybrid system.
- If the yeast cells co-transformed with the tested bait plasmid and empty pMyr or pMyr-Lamin C are able to grow at 36 °C, then it will be considered unsuitable for the CytoTrap two-hybrid system.
- The pSos bait plasmid needs to be co-transformed into yeast host cell cdc25H with negative control either pMyr or pMyr-Lamin C using YeastmakerTM Yeast Transformation System 2.
- DNA encoding bait protein is cloned into the pSos vector (plasmid map: http://www.chem-agilent.com/pdf/strata/217438.pdf) by the conventional enzyme-ligation method. The plasmid construct is confirmed by DNA sequencing to ensure the correct reading frame between hSos and bait.
- Generating a CytoTrap two-hybrid cDNA library
CytoTrap two-hybrid cDNA libraries can be constructed using mRNA from various organisms. Here we use the mRNA from the model plant Arabidopsis as an example. Briefly, mRNA is extracted and purified from leaf tissue of wild-type Arabidopsis plant (Col-0), then first-strand cDNA is synthesized using a hybrid oligo (dT) linker-primer that contains an XhoI restriction site and a high fidelity reverse transcriptase. 5-methyl dCTP is used in place of regular dCTP during the synthesis of first strand cDNA, and the methylated XhoI sites in the resulting cDNA will be resilient to XhoI digestion. The second-strand cDNA is synthesized using DNA polymerase I. An adapter containing a cohesive EcoRI end and a blunt end (see sequence in Note 1) is linked to the polished double stranded cDNA, and then the cDNA was digested with XhoI. The resulting cDNA containing EcoRI at 5’ end and XhoI at 3’ end is ligated into pMyr vector pre-cut by EcoRI and XhoI, so the cDNA is inserted in a sense expressional orientation driven by the Gal1 promoter (PGAL1). The cDNA library carried in pMyr plasmid (plasmid map: http://www.chem-agilent.com/pdf/strata/217438.pdf) is then transformed into high efficiency E. coli XL10-Gold Kan cells for propagation and storage. The detailed procedure is described below.
cDNA synthesis:
- mRNA purification. Leaf samples (~2 g) from 4-week-old Arabidopsis Col-0 plants are harvested, and total RNA is extracted from the leaf tissues using RNeasy Plant Mini Kit. Poly (A) mRNA is then purified from 1 mg of total RNA using Oligotex mRNA Mini Kit for cDNA synthesis (Zhang et al., 2014).
- First-strand cDNA synthesis is carried out using high fidelity reverse transcriptase AccuScript.
- Add the following reagents to an RNase-free microcentrifuge tube:
10x first-strand buffer: 5 μl
First-strand methyl nucleotide mixture: 3 μl
Linker-primer (1.4 μg/μl): 2 μl
RNase Block Ribonuclease Inhibitor (40 U/μl): 1 μl
mRNA: Equal to 5 μg
DEPC-treated water: Bring volume to 50 μl
- Incubate the reaction at room temperature for 10 min to allow primer to anneal to template.
- Add 3 μl of AccuScript reverse transcriptase to the reaction and incubate the reaction at 42 °C for 2 h.
- Add the following reagents to an RNase-free microcentrifuge tube:
- Second-strand cDNA synthesis.
- Put the first-strand cDNA reaction tube on ice, and add the following reagents to the tube:
10x second-strand buffer: 20 μl
Second-strand dNTP mixture: 6 μl
Sterile ddH2O: 139 μl
RNase H (5 U/μl): 1 μl
DNA polymerase I (9.0 U/μl): 11 μl
- Mix the reaction gently and spin down the reaction.
- Incubate at 16 °C for 3 h, and then put the tube on ice. The resulting double-strand cDNA (ds cDNA) should be ready for adapter ligation.
- Put the first-strand cDNA reaction tube on ice, and add the following reagents to the tube:
- Blunting cDNA termini and ligating the EcoR I adapter.
- Add the following reagents to second-strand cDNA synthesis, and incubate at 72 °C for 30 min.
10 mM dNTP mix: 4 μl
Phusion DNA polymerase (NEB): 4 μl
- Add 200 μl UltraPureTM phenol: chloroform: isoamyl alcohol (25:24:1) to the reaction and vortex to mix. Spin down the reaction at 14,000 x g at room temperature for 5 min, and then transfer the upper layer (~ 200 μl) to a new tube.
- Add 200 μl chloroform and vortex. Spin down the reaction at 14,000 x g at room temperature for 5 min, and then transfer the upper layer (~ 200 μl) to a new tube.
- Add the following reagents to precipitate cDNA, vortex and keep the reaction at -20 °C overnight.
3 M sodium acetate: 20 μl
100% (v/v) ethanol: 400 μl
- Spin down the cDNA at 14,000 x g for 60 min at 4 °C.
- Remove the supernatant carefully and wash the pellet using 70% ethanol (v/v). Spin down the pellet at 14,000 x g for 2 min at room temperature. Remove the ethanol and dry the pellet at room temperature.
- Resuspend the pellet in 8 ul EcoRI adapters and incubate at 4 °C for 1 h to ensure the complete dissolve of cDNA.
- Add the following reagents to the tube with cDNA and EcoRI adapters to bring volume to 10 μl, and incubate the reaction at 16 °C for overnight.
10x T4 ligase buffer (NEB): 1 μl
T4 ligase (NEB): 1 μl
- Incubate the tube at 70 °C for 30 min to terminate the reaction.
- Add the following reagents to second-strand cDNA synthesis, and incubate at 72 °C for 30 min.
- Phosphorylating the EcoRI end.
- Cool down the reaction at room temperature for 10 min.
- Add the following reagents to the tube, and incubate at 37 °C for 1 h.
10x reaction buffer (NEB): 1 μl
Sterile ddH2O: 8 μl
T4 polynucleotide kinase (NEB): 2 μl
- Incubate the tube at 70 °C for 30 min to terminate the reaction.
- Cool the reaction at room temperature for 10 min, and then add the following reagents to the reaction, incubate at 37 °C for 3 h.
10x reaction buffer (NEB): 5 μl
Sterile ddH2O: 22 μl
Xho I (NEB): 3 μl
- Precipitate the cDNA by adding the following reagents to the tube, and incubate at -20 °C overnight.
10x STE buffer: 5 μl
100% (v/v) ethanol: 125 μl
- Spin down the reaction at 14,000 x g for 60 min at 4 °C. Remove the supernatant and dry the pellet at room temperature. Dissolve the pellet in 100 μl sterile ddH2O.
- Cool down the reaction at room temperature for 10 min.
- Purify ds cDNA with CHROMA SPINTM+TE-400 columns. (Procedures are performed at room temperature if not specified.)
- Resuspend the gel matrix by invert the column several times. Remove the top cap and take away the break-away end at the bottom. Place the column in a 2-ml collection tube.
- Centrifuge at 700 x g for 5 min, and then remove the collection tube and buffer.
- Put the column to a new collection tube and add 100 μl cDNA sample to the center of top surface of gel matrix.
- Centrifuge at 700 x g for 5 min, collect the elution solution containing cDNA (size >200 bp).
- Precipitate cDNA by adding the following reagents to the eluted solution, and incubate at -20 °C freezer for overnight.
3 M sodium acetate (pH5.2): 10 μl
Ice-cold 100% ethanol: 250 μl
- Centrifuge at 14,000 x g for 30 min at 4 °C. Discard the supernatant, and add 360 μl 70% ethanol, inverted several times, and then centrifuge at 14,000 x g for 5 min at 4 °C. Remove the supernatant and dry the pellet at room temperature. Resuspend the cDNA in 5 μl sterile ddH2O.
- Resuspend the gel matrix by invert the column several times. Remove the top cap and take away the break-away end at the bottom. Place the column in a 2-ml collection tube.
- Ligating cDNA into pMyr vector and transform into E. coli.
- Set up T4 ligation reaction by adding the following reagents, incubate overnight at 16 °C.
10x ligase buffer (NEB): 2.5 μl
Resuspended cDNA (1 μg): X μl
pMyr vector (precut by EcoRI/XhoI, 500 ng): 5 μl
T4 DNA ligase (NEB): 2.5 μl
ddH2O: Bring volume to 25 μl
- Transform all the ligation reaction to XL10-Gold Kanr Ultracompetent cells. Use 2 μl ligation reaction for each transformation according to the manufacturer’s instruction.
- Plate the transformation on 150-mm LB plates containing chloramphenicol (25 μg/ml), about 50-100 plates in total. Spread appropriate volume of transformation, so that each plate may have 30,000 colonies. Incubate at 37 °C overnight to allow colonies to grow. The total number of colonies should be 3 million.
- Add 5 ml LB medium to each plate to scrape the bacteria with a spreader, and wash each plate using additional 1 ml LB medium. Pool all the bacteria in one beaker and mix gently and well. Aliquot 10 ml bacteria culture in 15-ml centrifuge tubes and stored at -80 °C. Purify plasmid from each 10 ml aliquot for CytoTrap two-hybrid screen.
- Set up T4 ligation reaction by adding the following reagents, incubate overnight at 16 °C.
- mRNA purification. Leaf samples (~2 g) from 4-week-old Arabidopsis Col-0 plants are harvested, and total RNA is extracted from the leaf tissues using RNeasy Plant Mini Kit. Poly (A) mRNA is then purified from 1 mg of total RNA using Oligotex mRNA Mini Kit for cDNA synthesis (Zhang et al., 2014).
- Screening a CytoTrap two-hybrid cDNA library
The bait and cDNA library plasmids are co-transformed into temperature sensitive host strain cdc25H. The transformation reaction is spread on SC/-LU plates containing glucose at room temperature to allow positive transformants to grow, and then the transformants are replica plated to SC/-LU plates containing galactose and incubated at 36 °C to select clones which expressing the bait and a potential interaction partner genes. The positive clones are further analyzed to confirm the bait-specific protein interaction. The flowchart of library screening is shown in Figure 1.
- Co-transformation of bait and cDNA library plasmids into host strain cdc25H.
This step is conducted as suggested by YeastmakerTM Yeast Transformation System 2 manual except that all the yeast culture steps must be conducted at room temperature unless otherwise specified to reduce rate of revert mutation of cdc25H. The transformation reactions are spread on SC/-LU plates (150-mm) containing glucose and kept at room temperature for 3 to 4 days until colonies grow to 2-3 mm in diameter. It is ideal to have ~3,000 colonies on each plate, and it is necessary to screen at least 1 million colonies.
- Replica plate the colonies grown up to SC/-LU plates (150-mm) containing galactose, and leave the plates at room temperature for one hour before moving to the selective temperature of 36 °C for incubation of 3 to 6 days. Colonies containing putative interacting partners and reverting mutants will grow up and it is necessary to pick up the colonies when they are 2-3 mm in diameter during selection, and not wait to pick up at the end of selection.
- Pick up colonies grown up on SC/-LU plates containing galactose at 36 °C, and dissolve the colonies in 25 μl sterile dH2O. Dot 5 μl of yeast suspension to four plates: Two SC/-LU containing glucose and two SC/-LU containing galactose. Incubate one SC/-LU plate containing glucose and one SC/LU plate containing galactose at room temperature, and the other two plates at 36 °C.
Notes:
- The number of plates should be scaled up if there are more yeast colonies.
- As shown in Figure 1, yeast colonies that only grow on SC/-LU containing galactose, but not on SC/LU containing glucose at 36 °C are kept as primary positive candidates for further analysis.
- The yeast colonies that grow on both galactose and glucose plates at 36 °C are temperature revert mutants, and their growth at selective temperature is caused by revert mutation of cdc25H mutant gene back to wild-type gene, hence their growth at 36 °C is not galactose-dependent.
- A large percentage of false positives will be the temperature revert mutants and can be easily identified by testing galactose-dependent growth at 36 °C.
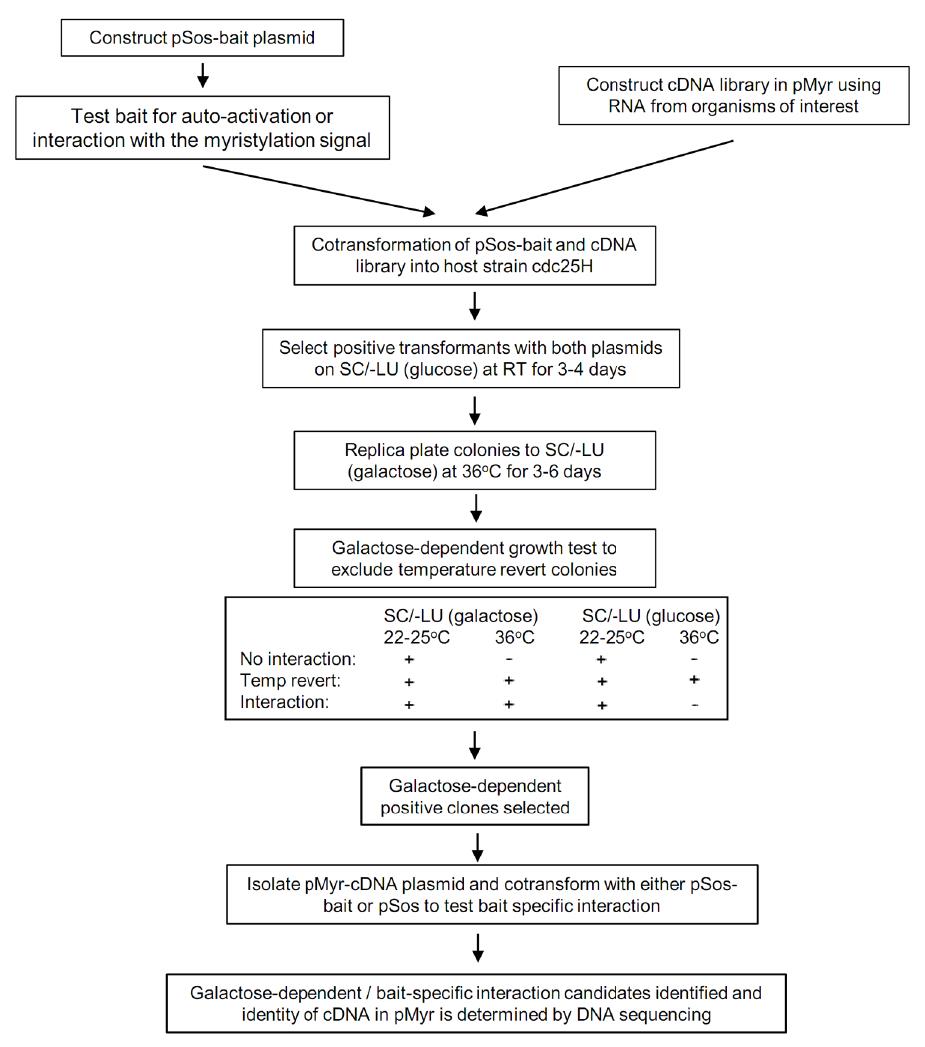
Figure 1. CytoTrap two-hybrid library screening procedure. A pSos-bait is co-transformed into yeast strain cdc25H with a cDNA library in pMyr. Positive transformants are selected on SC/-LU (glucose) plates at room temperature. Colonies are then replica plated on SC/-LU (galactose) plates and incubated at 36 °C for 3 to 6 days. Colonies grown up at 36 °C are further tested for galactose- and bait-specific growth to identify the true interaction clones.
- The number of plates should be scaled up if there are more yeast colonies.
- Co-transformation of bait and cDNA library plasmids into host strain cdc25H.
- Analyze positive interaction candidates
The primary positive candidates from library screen need to be further tested to see whether the interaction is bait-specific and nature of the prey genes need to be identified for functional analysis.
- Isolate prey plasmids from positive candidates.
- Inoculate positive yeast candidates in 2 ml of liquid SC/-LU containing glucose and shake at 30 °C for 2 days.
- Transfer 1.5 ml yeast culture to a 1.7-ml centrifuge tubes, and spin down the cells at 14, 000 x g for 1 min. Remove the supernatant and add 200 μl yeast lysis solution, resuspend cells by vortexing.
- Add 200 μl UltraPureTM phenol: chloroform: isoamyl alcohol (25:24:1, v/v) and 0.3 g of acid-washed glass beads, vortex vigorously for 2 min for each sample.
- Centrifuge at 14,000 x g for 5 min at room temperature, and transfer the top layer into a new microcentrifuge tube. Add the following reagents to each tube, and keep the tubes at -20 °C for 30 min.
3 M sodium acetate (pH 5.2): 20 μl
Ice-cold 100% ethanol: 500 μl
- Spin down the pellet at 14,000 x g for 10 min at 4 °C. Remove the supernatant, and wash the pellet with 70% ethanol. Spin down the pellet at 14,000 x g for 5 min at room temperature. Remove the supernatant and dry the pellet at room temperature.
- Resuspend the pellet in 50 μl sterile ddH2O, and then use 1-2 μl of yeast plasmid DNA to transform 10-beta Electrocompetent E. coli competent cells. Spread the transformation reaction on LB/chloramphenicol agar plates. Put at 37 °C for overnight.
- Pick up single colonies and inoculate in LB/chloramphenicol liquid medium at 37 °C for overnight. Purify the plasmid for further analysis.
- Inoculate positive yeast candidates in 2 ml of liquid SC/-LU containing glucose and shake at 30 °C for 2 days.
- Yeast co-transformation to test bait-specific protein-protein interaction
- Transform the purified candidate prey plasmids with either pSos-bait or pSos empty vector into host strain cdc25H.
- Inoculate the transformants on SC/-LU containing glucose at room temperature for 4 days. Pick up three individual colonies and transfer to SC/-LU containing glucose and SC/-LU containing galactose plates, and incubate at 36 °C for 3 to 6 days.
- Only prey plasmids that show specifically interact with pSos-bait, not with pSos empty vector are kept for sequence analysis.
- The identity of cDNA insert is determined by DNA sequencing.
- Transform the purified candidate prey plasmids with either pSos-bait or pSos empty vector into host strain cdc25H.
- Isolate prey plasmids from positive candidates.
Notes
- Sequence of adaptor containing a cohesive EcoR I end and a blunt end:
5´-OH-AATTCGGCACGAGG-3´
3´-GCCGTGCTCCp-5´
Recipes
- Yeast lysis solution (100 ml)
2 ml Triton® X-100
1 g SDS
0.58 g NaCl
1 ml of 1 M Tris-HCl (pH 8.0)
200 μl of 0.5 M EDTA (pH 8.0)
Bring volume to 100 ml using sterile dH2O
Stored at room temperature
- YPDA/YPAD (1 L)
10 g yeast extract
20 g Bacto® peptone
20 g dextrose
40 mg adenine sulfate
Bring volume to 1 L using sterile dH2O, and adjust to pH 5.7-5.8
If preparing plates, add 15 g agar
Autoclave at 121 °C for 20 min
- SC/-LU (glucose, 1 L)
6.7 g yeast nitrogen base without amino acids
20 g dextrose
0.6 g DO Supplement -His/-Leu/-Trp/-Ura
0.02 g histidine
0.05 g tryptophan
Bring volume to 1 L using sterile dH2O, and adjust to pH 5.7-5.8
If preparing plates, add 15 g agar
Autoclave at 121 °C for 20 min
- SC/-LU (galactose, 1 L)
6.7 g yeast nitrogen base without amino acids
20 g of galactose
10 g of raffinose
0.6 g DO supplement -His/-Leu/-Trp/-Ura
0.02 g histidine
0.05 g tryptophan
Bring volume to 1 L using sterile dH2O, and adjust to pH 5.7-5.8
If preparing plates, add 15 g agar
Autoclave at 121 °C for 20 min
- 10x STE buffer (1 L)
58.44 g NaCl
200 ml of 1 M Tris-HCl (pH 7.5)
200 ml of 0.5 M EDTA
Bring volume to 1 L using sterile dH2O
Acknowledgments
The research was supported by National Science Foundation grant 1021344 to B.W. Poovaiah, and the National Science Foundation of China grant U1130304 to Liqun Du. We thank Lorie Mochel and Ade Snider at Washington State University for their help in preparing the manuscript.
References
- Aronheim, A., Zandi, E., Hennemann, H., Elledge, S. J. and Karin, M. (1997). Isolation of an AP-1 repressor by a novel method for detecting protein-protein interactions. Mol Cell Biol 17(6): 3094-3102.
- Li, N., Batzer, A., Daly, R., Yajnik, V., Skolnik, E., Chardin, P., Bar-Sagi, D., Margolis, B. and Schlessinger, J. (1993). Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature 363(6424): 85-88.
- Zhang, L., Du, L., Shen, C., Yang, Y. and Poovaiah, B. W. (2014). Regulation of plant immunity through ubiquitin-mediated modulation of Ca(2+) -calmodulin-AtSR1/CAMTA3 signaling. Plant J 78(2): 269-281.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Zhang, L., Du, L. and Poovaiah, B. W. (2014). CytoTrap Two-Hybrid Screening Assay. Bio-protocol 4(23): e1310. DOI: 10.21769/BioProtoc.1310.
Category
Plant Science > Plant biochemistry > Protein > Interaction
Microbiology > Microbial genetics > DNA > DNA interaction
Molecular Biology > Protein > Protein-protein interaction
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.