- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Fluorescence Recovery after Photobleaching (FRAP) Assay to Measure the Dynamics of Fluorescence Tagged Proteins in Endoplasmic Reticulum Membranes of Plant Cells


Published: Vol 4, Iss 20, Oct 20, 2014 DOI: 10.21769/BioProtoc.1268 Views: 18767
Reviewed by: Hiromasa SaitohTie LiuArsalan Daudi
Original research article
The authors used this protocol in:

Mar 2014
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
In this protocol, we used fluorescence recovery after photobleaching (FRAP) to measure the influence that some mutations and drug treatment have on mobility of a green fluorescent protein (GFP)-fused viral transmembrane protein into endoplasmic reticulum membranes (Serra-Soriano et al., 2014). The proteins of interest were transiently expressed in Nicotiana benthamiana (N. benthamiana) epidermic cells by agro-infiltration. To minimize transient overexpression artifacts, fluorescence intensity values were gathered at 36 hpi using an inverted Zeiss LSM 780 confocal microscope. Only epidermic cells showing moderated expression levels and homogenous distribution through the ER of the GFP-tagged proteins were used for further experiments. To examine the role of actin polymerization in the mobilization of GFP-tagged proteins, we pretreated tissue samples either with latrunculin B, an inhibitor of actin polymerization, or with DMSO as control. The generated fluorescence recovery curves were used to obtain the percentage of maximum fluorescence recovery (MFR), which corresponds to the mobile fraction, and the half-time of maximum recovery (t1/2) values.
Materials and Reagents
- 3-4 weeks old Nicotiana benthamiana plants
- Agrobacterium tumefaciens (A. tumefaciens) C58C1 strain or similar transformed with the binary vector pMOG800 harboring the protein coding sequence. In our case they were:
- pMGFP-p7B encoding GFP-fused Melon necrotic spot virus (MNSV) p7B
- pMGFP-p7B [D7AP10A] encoding GFP-fused MNSV p7B carrying D7AP10A mutation
- pMGFP-p7B [K49A] encoding GFP-fused MNSV p7B carrying K49A mutation
- pMGFP-KDEL encoding a GFP engineered to be targeted to the ER lumen
- pMGFP-p7B encoding GFP-fused Melon necrotic spot virus (MNSV) p7B
- Yeast extract (Difco, catalog number: 212750 )
- Tryptone (Difco, catalog number: 211705 )
- Sodium chloride (NaCl) (Panreac Applichem, catalog number: 131659 )
- Acetosyringone (Sigma-Aldrich, catalog number: D134406 )
- MES (Sigma-Aldrich, catalog number: M8250 )
- Magnesium chloride (MgCl2) (Sigma-Aldrich, catalog number: M9272 )
- Antibiotics
- Mowiol® 4-88 (Sigma-Aldrich, catalog number: 81381 )
- Dabco® 33-LV (Sigma-Aldrich, catalog number: 290734 )
- Tris base (Roche Diagnostics, catalog number: 03118142001 )
- Tris-HCl (1 M, pH 5.6)
- Immersion oil Immersol 518 F (ZEISS, catalog number: 444960-0000-000 )
- Latrunculin B (Sigma-Aldrich, catalog number: L5288 )
- 25 µM Latrunculin B solution in 10 mM DMSO
- LB medium (see Recipes)
- Agrobacterium infiltration buffer (see Recipes)
- Mowiol mounting medium (see Recipes)
Equipment
- 28 °C growing chamber
- 15 ml culture tubes
- Swinging centrifuge rotor for 15 ml tubes
- 1.5 ml tubes (standard Eppendorf tubes or similar)
- BioPhotometer plus (Eppendorf)
- 1 ml syringes without needle
- Plant growing chamber
- Fine paddle forceps
- Petri culture dishes 35 x 10 mm
- Microscope slides 76 x 26 mm (Menzel-Gläser, catalog number: AA00000112E )
- Microscope cover slips 24 x 24 mm Nr. 1 (Menzel-Gläser, catalog number: BB024024A1 )
- Adhesive one-sided tape
- A cork borer (one centimeter diameter) or similar cutting device such as a scalpel blade.
- An inverted confocal microscope (ZEISS, model: LSM 780)
- Plan-Apochromat 63x/1.40 oil objective
Software
- Graphpad prism software (http://www.graphpad.com)
- MS Excel
Procedure
- Transient expression of proteins by agro-infiltration
- Inoculate a single colony of transformed A. tumefaciens in 5 ml of LB medium with Kanamycin (50 µg/ml) and Rifampicin (50 µg/ml). Incubate the culture (12 h or more) at 28-30 °C with vigorous shaking.
- Measure the A600 of the culture.
- Collect the bacteria by slow-speed centrifugation (2,000 x g, 15 min) in a swinging rotor. Remove the supernatant.
- Resuspend the pellet with Agrobacterium infiltration buffer. The final A600 should be adjusted to 0.2.
- Leave at room temperature for 2 h before infiltration.
- Infiltrate the cultures with 1 ml syringe into the lower side of the leaves. Use 3-4 week-old N. benthamiana plants and avoid cotyledons. Use your fingertip to apply gentle counter pressure to the other side of the leaf. A fully infiltrated part of the leaf gives a water-soaked appearance.
- Keep the plants in growth chambers in 16 h light at 25 °C and 8 h dark at 22 °C.
- Inoculate a single colony of transformed A. tumefaciens in 5 ml of LB medium with Kanamycin (50 µg/ml) and Rifampicin (50 µg/ml). Incubate the culture (12 h or more) at 28-30 °C with vigorous shaking.
- Microscope slide preparation
- To minimize transient overexpression artifacts in the FRAP experiments plants were used 36 h after infiltration.
- Cut a small disk of the infiltrated leaf with a cork borer or similar device. Try to avoid the leaf veins to eliminate any potential irregularity or significant depression in the sample.
- In the case of drug treatment, immerse tissue samples into latrunculin B (1 ml of a 25 µM solution) in a small petri culture dish (35 x 10 mm) for 1h. Perform non-treated control by tissue immersion into 10 mM DMSO.
- Using fine paddle forceps place the leaf disk in a microscope slide with the back side facing up and mount in Mowiol mounting medium.
- Add a coverslip and stick them to the slide with adhesive one-sided tape.
- Place a drop of immersion oil on the cover slip over the leaf disk.
- To minimize transient overexpression artifacts in the FRAP experiments plants were used 36 h after infiltration.
- Fluorescence recovery after photobleaching (FRAP) assay
- FRAP experiments were performed on an inverted Zeiss LSM 780 confocal microscope using the LSM FRAP Module from Zen 2011 software.
- Switch on the laser lines for imaging the sample.
- Find epidermic cells showing moderated expression levels and homogenous distribution through the ER of the GFP-tagged proteins with the Plan-Apochromat 40x/1.4 oil objective and then switch to the Plan-Apochromat 63x/1.40 oil objective to capture images.
- Select “range indicator”. Now image turn into one of two-color (red and blue) (Figure 1). The red areas indicated saturation of the detector due to high fluorescence intensity. Similarly, the blue area either represents no fluorescence or intensity lower than the detector can sense. Only a few pixels have to be saturated (in red), if not, adjust the pinhole, laser intensity, or detector gain.
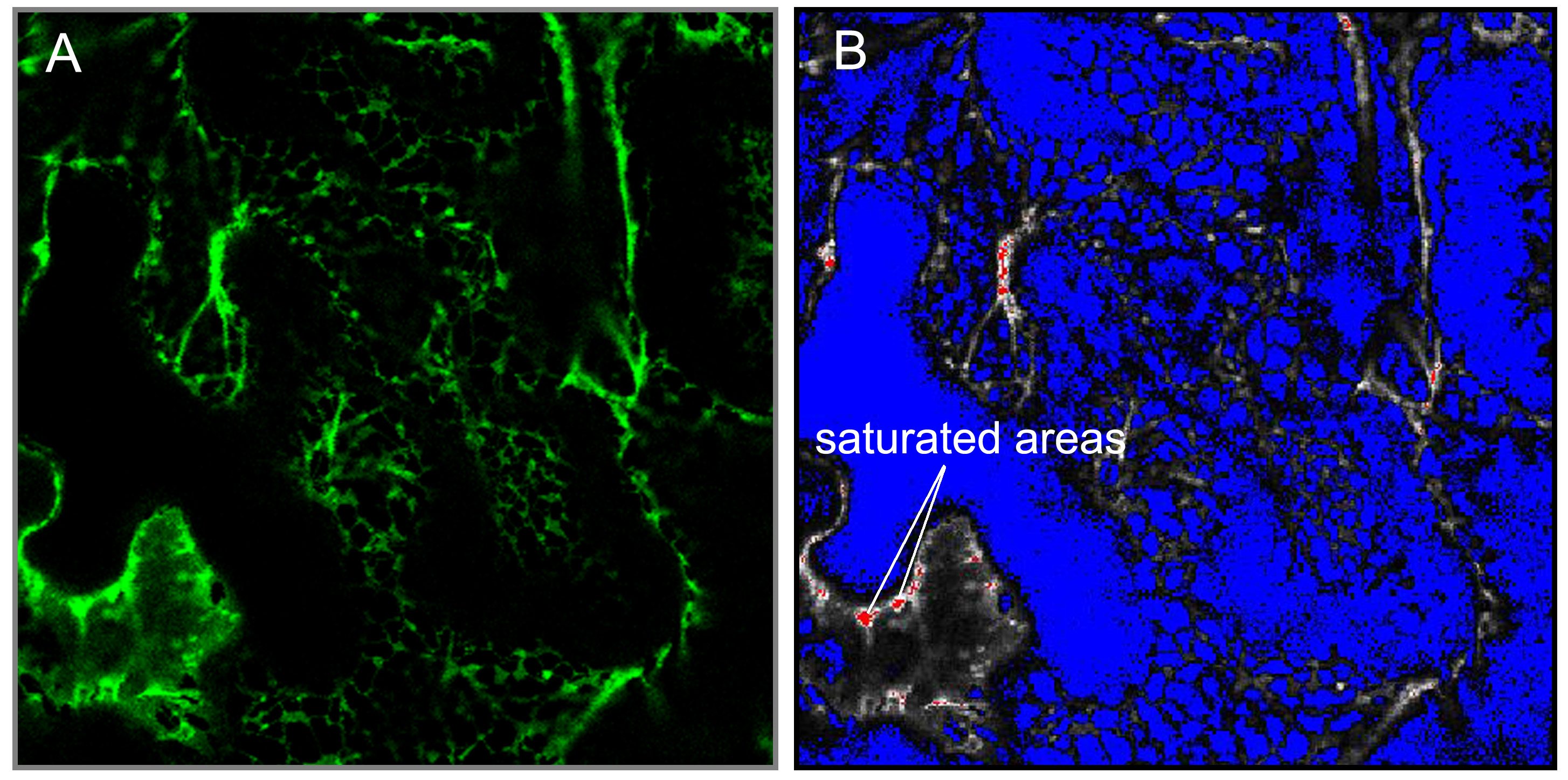
Figure 1. Laser confocal scan in green A and range indicator mode B. The saturated areas in red are indicated. - Use 5x optical zoom and 256 x 256 pixel resolution to take a single image of a short piece of the endoplasmic reticulum (for this purpose use “Snap” function). This zoom factor should stay the same between samples.
- Click the bleaching option on the acquisition tab. Time series and region options will be automatically selected.
- To define the regions of interest (ROI) which should be bleached select the circular shape in the regions menu and draw four circular ROIs of 1.7 μm diameter in fluorescent regions of the sample. Select a flat membrane region of the ER significantly larger than the bleach ROIs. Three of ROIs will be bleached and the fourth one will be used as the non-bleached reference to correct the FRAP curves for photofading caused by imaging. Locate the reference ROI as far apart as possible from the bleached ROIs. Ensure that a region which is background is in your image and draw a fifth circular ROI inside. In fast recovery rates, it is important to use small ROIs to decrease at maximum the time interval between photobleaching and imaging.
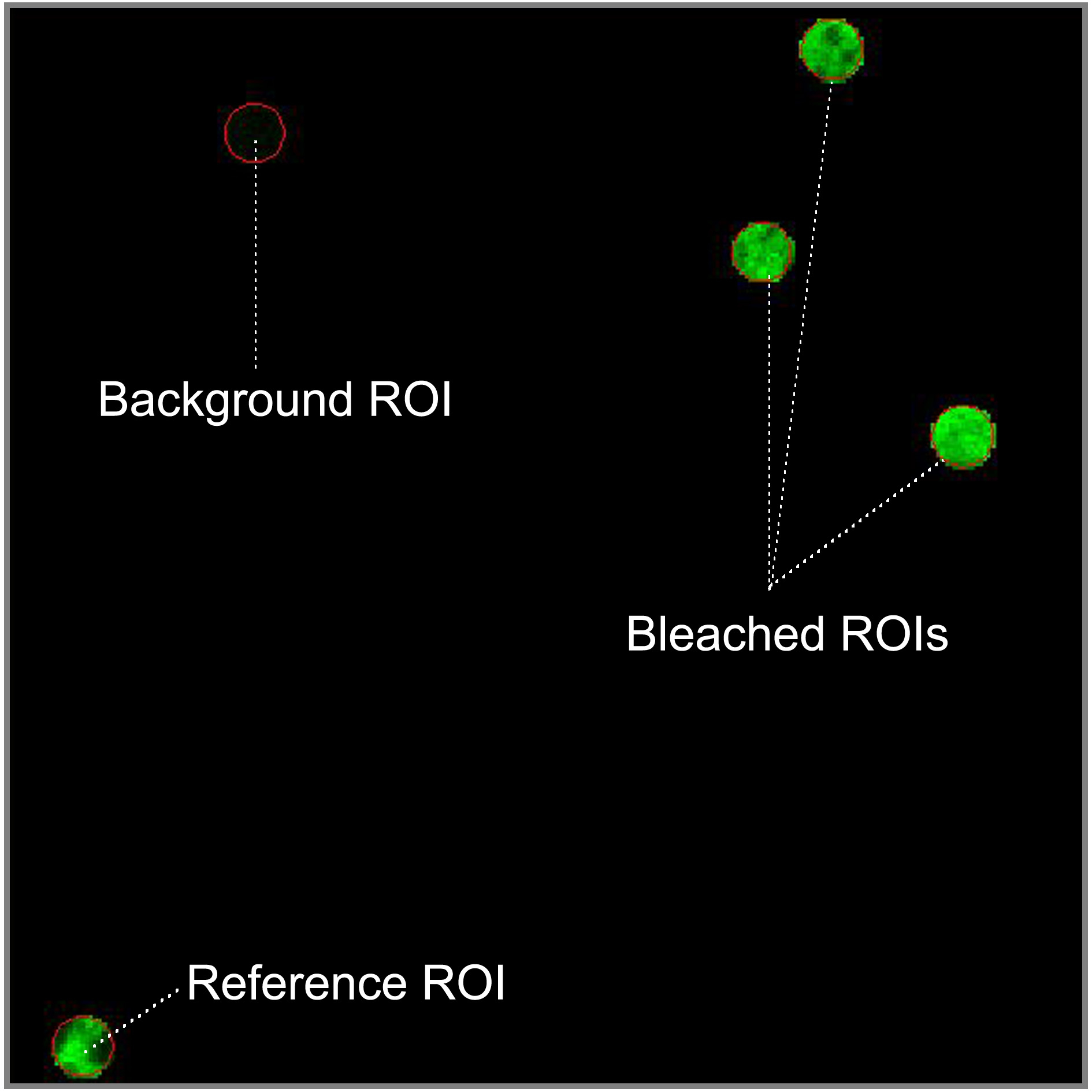
Figure 2. Spatial distribution of the región of interest (ROIs). Reference ROI must be located away from bleached ROIs to be not affected by the laser bleach. Background ROI should be in a non-fluorescent area (blue pixels in range indicator mode). - To setup bleaching protocol select start bleaching after 3 scans and use 100 iterations of the bleach laser over the bleach region. Do not use zoom bleach. In excitation of bleach, select the 488 nm line of the Argon laser (25% power, 100% transmission). After bleaching, try to reduce the ROI intensity between 50-75% without photodamage to the cell. After bleaching collect images at low laser power (25% power, 2% transmission).
- Open the time series menu and specify the total number of images in the final data set (before and after bleaching) by setting the number of cycles to 100. This results in an interval between the beginning of each scan of approximately 191 ms. To further increase the rate of data acquisition, use bidirectional scanning.
- Start FRAP experiment by pressing “Start Experiment button” and examine the FRAP curve on the computer. ROI reference intensity should remain constant or be slightly reduced due to photofading during image acquisition Repeat FRAP on different cells and days to ensure reproducibility. To allow comparison between different cells and proteins, the detector gain can be readjusted but all other settings must be kept the same.
- FRAP experiments were performed on an inverted Zeiss LSM 780 confocal microscope using the LSM FRAP Module from Zen 2011 software.
- FRAP data processing
- FRAP data generated using the LSM systems can be displayed and analyzed with the confocal operating software. Alternatively, raw fluorescence intensity values from bleached, background and reference ROIs can be exported to Microsoft Excel to be double normalized (Phair et al., 2004).
- Calculate the photobleaching rate (R) by comparing the intensity of the fluorescence of the reference ROI before (RF0, average intensity of the three scans taken before bleaching) and at each scan after photobleaching (RFn) Rn = Fn/F0.
- Normalize the fluorescence intensity of the bleached ROIs (BleachedFn) as follows: NF = (BleachedFn - BackgroundFn)/r.
- Copy the data to a program such as GraphPad Prism and curve fit the fluorescence by using non-linear regression and the exponential one-phase association model. The mobile fraction (Mf) that corresponds to the plateau value and half-life
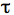 1/2, the time it takes for fluorescence intensity to reach half the maximum of the plateau level are obtained by the software.
1/2, the time it takes for fluorescence intensity to reach half the maximum of the plateau level are obtained by the software.
- FRAP data generated using the LSM systems can be displayed and analyzed with the confocal operating software. Alternatively, raw fluorescence intensity values from bleached, background and reference ROIs can be exported to Microsoft Excel to be double normalized (Phair et al., 2004).
Notes
- Every FRAP protocol need to be adapted depending on the nature of sample and preparation. FRAP setup shown herein is merely orientative and should not be considered as guarantee of good results. Then, it is very likely your first session efforts will be focused in optimizing photobleaching and data acquisition (Carisey et al., 2011).
Recipes
- LB medium
Mix 5 g of yeast extract with 10 g of tryptone, 10 g of NaCl
Add dH2O to 1,000 ml - Agrobacterium infiltration buffer
10 mM MES (pH 5.6)
10 mM MgCl2
150 µM acetosyringone - Mowiol mounting medium
- Add 9.6 ml of Mowiol 4-88 to 24 ml of glycerol and 24 ml of dH2O.
- Stir to mix and leave for several hours at room temperature.
- Add 9.6 ml of 1 M Tris-Cl (pH 8.0) and 38.4 ml of dH2O heat to 50 °C for 10 min with occasional mixing.
- After the Mowiol dissolves (approximately 3 h), clarify by centrifugation at 5,000 x g for 15 min.
- For fluorescence detection, add Dabco to 2.5% to reduce fading.
- Aliquot in 1.5 ml tubes and stored at -20 °C.
- Add 9.6 ml of Mowiol 4-88 to 24 ml of glycerol and 24 ml of dH2O.
Acknowledgments
This work was funded by grant BIO2011-25018 from the Spanish Ministerio de Economia y Competitividad and by Prometeo Program GV2011/003 from the Generalitat Valenciana. J.A.N. and M.S. are the recipients of a postdoctoral contract and a PhD fellowship from the Ministerio de Educacion y Ciencia of Spain.
References
- Carisey, A., Stroud, M., Tsang, R. and Ballestrem, C. (2011). Fluorescence recovery after photobleaching. Methods Mol Biol 769: 387-402.
- Phair, R. D., Gorski, S. A. and Misteli, T. (2004). Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy. Methods Enzymol 375: 393-414.
- Serra-Soriano, M., Pallas, V. and Navarro, J. A. (2014). A model for transport of a viral membrane protein through the early secretory pathway: minimal sequence and endoplasmic reticulum lateral mobility requirements. Plant J 77(6): 863-879.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Navarro, J. A., Serra-Soriano, M. and Pallás, V. (2014). Fluorescence Recovery after Photobleaching (FRAP) Assay to Measure the Dynamics of Fluorescence Tagged Proteins in Endoplasmic Reticulum Membranes of Plant Cells. Bio-protocol 4(20): e1268. DOI: 10.21769/BioProtoc.1268.
Category
Plant Science > Plant cell biology > Cell imaging
Cell Biology > Cell imaging > Live-cell imaging
Cell Biology > Cell imaging > Fluorescence
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.