- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Lysosomal Stability Assay
Published: Vol 4, Iss 12, Jun 20, 2014 DOI: 10.21769/BioProtoc.1162 Views: 18193
Reviewed by: Lin Fang
Original research article
The authors used this protocol in:

Sep 2013

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics






Abstract
This assay makes use of the dye Acridine Orange (AO) to determine the stability of lysosomes in living cells upon exposure to a confocal microscope laser.
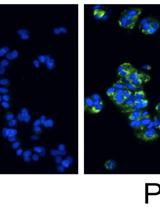
AO is a lipophilic amine that readily diffuses into cells (Figure 1). Inside the cell it enters the acidic lysosomal compartment where it is protonated and sequestered, shifting its emission spectrum towards a longer wavelength (i.e. red). Once inside the lysosomes, the metachromatic AO sensitizes the lysosomal membrane to photo-oxidation by blue light (Brunk et al., 1997). Upon light-induced loss of the lysosomal pH gradient and subsequent leakage of AO into the cytosol, the emission spectrum of AO shifts from red to green (Figure 2). Hence, loss of lysosomal integrity can be measured as a ‘loss of red dots’ or as a quantitative rise in green fluorescence (Petersen et al., 2010; Kirkegaard et al., 2010; Petersen et al., 2013).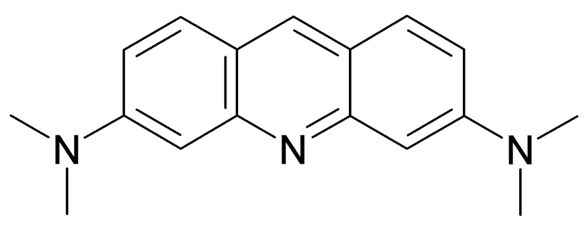
Figure 1. Acridine Orange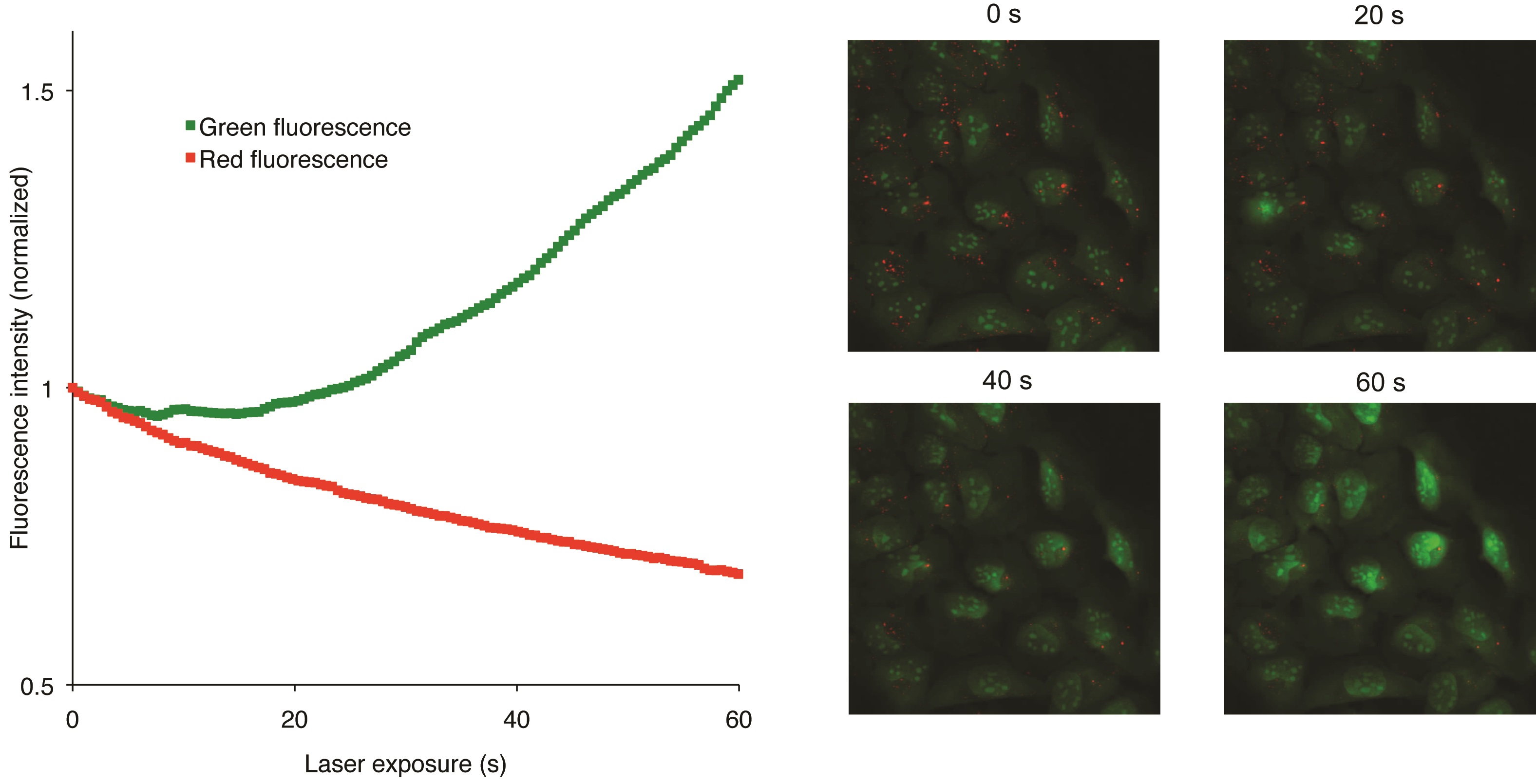
Figure 2. Snapshots visualizing the U2OS cells at various steps of the recording procedure (Petersen et al., 2010)
Materials and Reagents
- Cells (such as U-2 OS, ATCC, catalog number: HTB-96)
- Growth medium (such as RPMI 1640 with 6% serum, Life Technologies, Gibco®)
- Acridine Orange (Sigma-Aldrich, catalog number: 235474)
- PBS containing 3% serum
Equipment
- Zeiss Live DUO 5 confocal microscope or any newer microscope model capable of recording confocal images at high speed (500 ms/exposure), 100 mW diode laser, 40x c-apochromat objective
- Lab-Tek 4 well (Nunc®, catalog number: 155383 ) or 8 well (Nunc®, catalog number: 155411 ) borosilicate cell chambers
Procedure
Notes on protocol versions: This protocol exists in two versions, one for use with common cancer cell lines (such as U2OS or Mcf7) and one for mouse embryonic fibroblasts (MEFs) transduced with either the SrcY527F oncogene or empty vector (or indeed any other transduced genes of interest). The main difference between the two protocols is in the recording of the images on the confocal microscope. When needed, both versions will be described at each step and designated either COMMON or MEF.
- Grow cells in borosilicate lab-tek wells overnight or for as long as required by necessary treatments. The exact nature of the growth medium is not important. The amount of cells in each well on the day of analysis is critical to the successful use of this protocol. In order to keep the variation low, the cells should be 50-75% confluent, and should never reach 100% confluency.
- Add Acridine Orange directly to the growth medium to a final concentration of 2 µg/ml. Incubate the cells at 37 ºC for 20 min.
- Wash the cells twice in PBS containing 3% serum.
- Add PBS with 3% serum to cover the cells (100 or 200 µl for 8-well or 4-well respectively).
- Proceed immediately to analysis at the microscope. The cells are only suited for analysis for the first 60 min after washing. After this time, the variation between different observation fields will increase.
- Identify a group of cells in the brightfield, and make sure the lysosomes (visible as black dots) are in focus. Be quick at this point, since brightfield also contains blue light.
- Microscope settings:
This protocol was developed and used on a Zeiss LSM 5 LIVE DUO microscope. It is important to note that, as for all microscope protocols, the exact settings should be determined empirically for each instrument and experimental setup. As noted above, the protocol used for ‘normal’ cancer cell lines (i.e. U2OS & Mcf7) was different from the one used for MEFs. The slower protocol with less frames (and less exposure to blue light) for MEFs was developed since lysosomes loose their integrity a lot faster in these cells, making quantification challenging when recorded at high speed.Heat chamber 37 ºC Ocular 40x c-apochromat Laser intensity 2.6 (most cancer cell lines)/0.9 (MEF) (can vary greatly between instruments) Image quality 8 bit (16 bit makes the files unnecessary large) Recording speed 1 frame every 0.5 (most cancer cell lines)/12 sec (MEF) Images recorded 200 (most cancer cell lines)/120 (MEF) Laser wavelength 489 nm Recording filters 495-555 nm band pass (green), 650 nm long pass (red) - Record movies. Any focus corrections should be made within the first 10 sec of recording. Make sure never to record the same place twice (easy to see due to absence of red dots). Allow time for all wells to be analyzed within an hour. Note the order in which the recordings were made, for use in troubleshooting later.
Data analysis
The data was analyzed using the LSM5 DUO software. This software has since been upgraded by Zeiss to various versions of ZEN. Unfortunately a key functionality for this analysis was omitted in this update, so data analysis was performed in the older software. Hopefully this will be rectified by Zeiss, and maybe other microscope and software suppliers don’t have these issues.
- Open the movie in the Zeiss software.
- Choose ROI (region of interest) analysis.
- Determine the minimum pixel intensity to be analyzed for each channel (or colour). This is done using the “level” function. At the time of writing, the ZEN software could only do this on single images, not the multiple image frames making up the movies recorded in this protocol. The older Zeiss software can do this.
- Forward the movie to the first frame after 10 sec.
- Mark the entire frame with the ROI tool. In previous versions of this protocol individual cells were marked manually, however this was very labour intensive, and will not produce better results.
- For the green channel, change the minimum level of pixel intensity to be analyzed, so that all background is omitted (The entire area of cells should be visible with a weak green stain.).
- For the red channel, change the level so that only brightly red stained lysosomes are analyzed.
- These same numeric settings should be used for all movies.
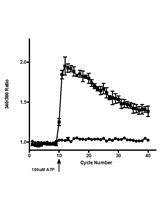
- Since each frame contains multiple cells, the values calculated by the software should now reflect the average green and red intensity of the cells. Observe that the green values increase while the red values decrease as the lysosomes burst.
- Copy the values from the microscope software directly to Excel or a similar program.
- Forward the movie to the first frame after 10 sec.
- In excel, line up data sets that come from identical experimental settings. At least three independent experiments, each with 3-4 movies, should be analyzed.
- Normalize each movie to its value at 10 sec.
- Visualize the increase in green in a x/y scatter plot. The decrease in red is much less reliable than the increase in green. Warning, this produces large and complex spreadsheets (Figure 3).
- For statistical evaluation, a simple students t-test can be performed for individual time points (for example the control value at 20 sec versus the treated value at 20 sec).
Figure 3. Example of data set in Excel
Notes
It is important to emphasize that great care should be taken to standardize the cell growth conditions, as the outcome of this assay is greatly affected by confluency and the general health status of the cells. Also, lipid mediated transfection protocols can influence the integrity of lysosomes negatively, making them more unstable. Finally, it is expected that at the end of each movie, the intensity of the green staining will start to decrease towards the end of each movie, so when showing graphs it is often required to omit the late values for clarity.
Acknowledgments
This protocol was developed for two papers addressing the stability of lysosomes (Petersen et al., 2013 and Kirkegaard et al., 2010). The work was supported by grants from the Danish Cancer Society (to NHTP and MJ), and the Danish National Research Foundation, the Danish Council for Independent Research, the European Commission FP7 and the Novo Nordisk Foundation (to MJ).
References
- Brunk, U. T., Dalen, H., Roberg, K. and Hellquist, H. B. (1997). Photo-oxidative disruption of lysosomal membranes causes apoptosis of cultured human fibroblasts. Free Radic Biol Med 23(4): 616-626.
- Kirkegaard, T., Roth, A. G., Petersen, N. H., Mahalka, A. K., Olsen, O. D., Moilanen, I., Zylicz, A., Knudsen, J., Sandhoff, K., Arenz, C., Kinnunen, P. K., Nylandsted, J. and Jäättelä, M. (2010). Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature 463(7280): 549-553.
- Petersen, N. H., Kirkegaard, T., Olsen, O. D. and Jäättelä, M. (2010). Connecting Hsp70, sphingolipid metabolism and lysosomal stability. Cell Cycle 9(12): 2305-2309.
- Petersen, N. H., Olsen, O. D., Groth-Pedersen, L., Ellegaard, A. M., Bilgin, M., Redmer, S., Ostenfeld, M. S., Ulanet, D., Dovmark, T. H., Lonborg, A., Vindelov, S. D., Hanahan, D., Arenz, C., Ejsing, C. S., Kirkegaard, T., Rohde, M., Nylandsted, J. and Jäättelä, M. (2013). Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell 24(3): 379-393.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Petersen, N. H. T., Kirkegaard, T. and Jäättelä, M. (2014). Lysosomal Stability Assay. Bio-protocol 4(12): e1162. DOI: 10.21769/BioProtoc.1162.
Category
Cancer Biology > General technique > Cell biology assays > Metabolism
Cell Biology > Cell staining > Organelle
Cell Biology > Cell imaging > Live-cell imaging
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.