- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Generation of Non-typeable Haemophilus influenzae Directed Gene Deletion Mutants

Published: Vol 4, Iss 5, Mar 5, 2014 DOI: 10.21769/BioProtoc.1066 Views: 11028
Reviewed by: Fanglian He
Original research article
The authors used this protocol in:

Jun 2013
Advertisement


Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics







Abstract
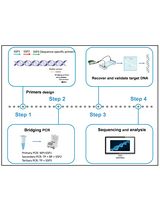
Directed deletion mutants in non-typeable Haemophilus influenzae can be made by allelic exchange of the target gene with an artificial DNA construct in which an antibiotic resistance cassette is placed between two ~1,000 bp DNA sequences that are identical to the 5' and 3' flanking regions of the target gene. The artificial DNA construct that is required for this mutagenesis is synthesized by the so-called Megaprimer PCR method (Figure 1).

Figure 1. Schematic representation of the Megaprimer PCR method. Step A1. The flanking regions (~1,000 bp) of the target gene are amplified by PCR (Primer pair L1 + L2 and R1 + R2). The L2 and R2 primers have extensions with homology to the antibiotic resistance cassette. Step A2. The antibiotic resistance cassette is amplified by PCR (primer pair L + R). Step B. The three PCR products of the first PCR reactions are combined. The flanking regions anneal to the antibiotic resistance cassette and one large PCR product is formed.
Materials and Reagents
- Amplitaq DNA polymerase including 25 mM MgCl2 and 10x reaction buffer (Applied Biosystems®, catalog number: N8080171 )
- PWO DNA polymerase including 25 mM MgSO4 and 10x reaction buffer (Roche Diagnostics, catalog number: 11644955001 )
- 10 mM dNTP mix
- Sterile Milli-Q water
- QIAquick PCR purification kit (QIAGEN, catalog number: 28104 )
- Agarose
- Ethidium bromide
- 0.5x TBE
- 100 bp perfect DNA ladder (Merck Millipore, catalog number: 70539 )
- 1 kb perfect DNA ladder (Merck Millipore, catalog number: 70537 )
- 1x PBS
- Bacto-agar (BD Biosciences, catalog number: 212030 )
- Bacto-Brain Heart Infusion medium (BD Biosciences, catalog number: 237500 )
- Glycerol
- M-IV competent non-typeable Haemophilus influenzae (Herriott et al., 1970), for protocol see below.
Equipment
- T100 thermal cycler (Bio-Rad Laboratories)
- Nanodrop spectrophotometer (Thermo Fisher Scientific, Nanodrop, model: ND1000 )
- Centrifuge (Eppendorf, model: 5810 )
- Microcentrifuge (Eppendorf, model: 5417R )
- Shaker (New Brunswick Scientific, model: Innova 4000 )
- 37 °C, 5% CO2 incubator (BINDER GmbH, model: CB 150 )
- DNA gel electrophoresis equipment
Procedure
- PCR amplify the antibiotic resistance cassette and the flanking regions of the target gene.
Please see Figure 1 about primer pairs, L1/L2, R1/R2 and L/R used in this PCR.
Reaction mix:
5 μl 10x PWO polymerase buffer (-MgSO4)
3 μl 25 mM MgSO4*
5 μl 2.5 mM dNTPs
5 μl 4 pmol/μl primer L1, R1, or L, respectively
5 μl 4 pmol/μl primer L2, R2, or R, respectively
1 μl 5 ng/μl template DNA*#
0.5 μl PWO DNA polymerase
25.5 μl dH2O
Vtot = 50 μl
PCR cycle:
93 °C- 4 min
93 °C30 sec |
55 °C1 min | 35x
68 °C1.5 min|
68 °C5 min
12 °C∞
* Optimize if necessary. Increased MgSO4 or DNA template concentrations might increase yield, but might also decrease specificity.
# Template for the flanking regions is chromosomal DNA. Template for the antibiotic resistance cassette can be plasmids or chromosomal DNA of a resistant strain.
- Purify PCR products with QIAquick PCR Purification Kit.
- Measure DNA concentration with the Nanodrop. Concentration of >20 ng/μl is enough for the Megaprimer PCR (step 6).
- Load 2 μl PCR product mixed with DNA loading buffer on a 1.5% agarose gel with a 100 bp DNA marker.
- Check the gel for purity (should be a single PCR product) and size of the product (size depends on the primer design, usually ~1,000 bp). When the desired product is not amplified, repeat the PCR in step 1 with modified conditions (e.g. decreased Tm, increased MgSO4 or addition of DMSO).
- Perform a MegaPrimer PCR to unite the left and right flanking region of the target gene with the antibiotic resistance cassette.
Reaction mix:
5 μl 10x PWO polymerase buffer (+20 mM MgSO4)
5 μl 2.5 mM dNTPs
~200 ng PCR product L1+ L2 (left flanking region)
~200 ng PCR product R1+ R2 (right flanking region
~400 ng PCR product R + L (antibiotic resistance cassette)
0.5 μl PWO DNA polymerase
Vtot = 50 μl with dH2O
PCR cycle:
93 °C 4 min
93 °C30 sec |
55 °C1 min | 30x
68 °C3 min |
68 °C7 min
12 °C∞
- Load 2 μl PCR product mixed with DNA loading buffer on a 0.7% agarose gel with a 1 kb DNA marker.
- Check the gel for purity and size of the PCR product. PCR products with multiple sizes can be present, but should contain a product with the size of the left flank, antibiotic cassette and right flank combined, typically ~3,000 bp.
- Optional: Perform a MegaPrimer PCR with one the PCR product of one flank and the antibiotic resistance cassette to confirm that the desired MegaPrimer PCR product is not formed without all three components.
- Optional: Improve PCR yield by adding primers L1 and R1 (see Figure 1) to the PCR mix.
- Thaw 1 ml M-IV competent NTHi (protocol for making competent M-IV see Reference 2 and protocol in Notes below).
- Centrifuge 2 min at 10,000 x g in microcentrifuge.
- Remove the M-IV medium containing glycerol and resuspend the bacteria in 1 ml M-IV medium.
- Add target DNA and incubate 100 min. shaking with 100 rpm at 37 °C.
- Centrifuge 2 min at 10,000 x g in microcentrifuge.
- Remove 900 μl M-IV medium and plate 100 μl on selective BHI plates. Grow overnight at 37 °C + 5% CO2.
Note: Record in your logbook the amount of colonies appearing on the selective plate.
- Pick four single colonies using a loop and streak them independently onto new selective BHI plates. Grow them overnight at 37 °C + 5% CO2.
Note: From now on this protocol until step 23 is done for all 4 colonies from step 17.
- Resuspend colonies from step 17 in 5 ml of BHI medium with antibiotics.
- Grow bacteria until optical density at 620 nm = 0.2.
- Perform a chromosomal DNA isolation on the 5 ml culture from step 19 (e.g. CTAB protocol, for protocol see Notes below).
- Perform two control PCRs with isolated genomic DNA from step 20 to confirm the deletion of the target gene and the insertion of the antibiotic resistance cassette. Include as negative control the chromosomal DNA of the wild type strain.
Reaction mix:
2 μl 10x AmpliTaq PCR buffer (-MgCl2)
2 μl 25 mM MgCl2*
2 μl 2.5 mM dNTPs
2 μl 4 pmol/μl primer L1
2 μl 4 pmol/μl primer C (gene) or primer A (cassette)
1 μl 5 ng/μl template chromosomal DNA
0.2 μl AmpliTaq DNA polymerase
8.8 μl dH2O
Vtot = 20 μl
PCR cycle:
93 °C4 min
93 °C30 sec |
55 °C1 min | 35x
72 °C1.5 min |
72 °C4 min
4 °C∞
*Optimize if necessary
- Load 2 μl PCR product mixed with DNA loading buffer on a 1.5% agarose gel with a 100 bp DNA marker.
- Check the gel for the size of the PCR product and select a mutant to continue with. Primer L1 + primer C (gene) should not result in an amplified PCR product (gene is removed), whereas the primer L1 + primer A (cassette) should give a PCR product (antibiotic cassette is inserted). Select the correct mutant and throw away all others.
- 1 µg chromosomal DNA of the mutant obtained in step 20 is 'crossed back' to the recipient wild type strain by transformation to prevent accumulation of (spontaneous) mutations because the chance that multiple mutations will transfer to the recipient bacterium is very low.
- Optional: To ensure that both mutant as wild-type strain have undergone the same (stress) conditions, also perform a transformation without adding DNA and without selection.
- Plate transformation mixture on (selective) BHI plates. Grow them overnight at 37 °C + 5% CO2.
- Optional: Make sure to dilute the wild type cells enough (e.g. 10,000 times) and plate onto a BHI agar plate without antibiotic. The mutant and equally treated wild type strain can be 'coupled' to for downstream experiments. It is possible to combine multiple mutants to one wild type strain.
- Pick a single colony from each plate using a loop and streak them independently onto new (selective) BHI plates. Grow them overnight at 37 °C + 5% CO2.
Note: From now on this protocol is identical for the wild type and mutant strains.
- Resuspend colonies from step 28 in 5 ml of BHI medium with or without antibiotics.
- Grow bacteria until OD 0.2 at 620 nm and place on ice.
- Pipet 1.0 ml into a cryotube with 0.25 ml 80% glycerol and store at -80 °C.
- Use of the remaining media for chromosomal DNA isolation.
- Perform a chromosomal DNA isolation on the 5 ml culture from step 19 (e.g. CTAB protocol, for protocol see Notes below).
- Perform two control PCRs with isolated genomic DNA from step 33 with similar conditions as in step 21 to confirm the deletion of the target gene and the insertion of the antibiotic resistance cassette. Include as negative control the chromosomal DNA of the wild type strain.
- Load 2 μl PCR product mixed with DNA loading buffer on a 1.5% agarose gel with a 100 bp DNA marker.
- Check the gel for the size of the PCR product and select a mutant to continue with. Primer L1 + primer C (gene) should not result in an amplified PCR product, whereas the primer L1 + primer A (cassette) should give a PCR product. Select the correct mutant and throw away all others.
Notes
- Generation of M-IV competent NTHi (for details see Reference 2)
- Inoculate 10-15 ml sBHI medium in a 50 ml tube with a few colonies of an overnight sBHI plate and grow to an OD600 = 0.30-0.35 with 200 rpm at 37 °C.
- Centrifuge 10 min with 3,000 x g at room temperature.
- Aspirate the sBHI medium and remove the last amount of sBHI medium with a p1,000 pipet.
- Resuspend the bacteria in 1 ml PBS and add PBS to the start volume.
- Centrifuge 10 min with 3,000 x g at room temperature.
- Aspirate the PBS and remove the last amount of PBS with a pipet.
- Resuspend the bacteria in 1 ml M-IV medium and add M-IV medium to 10-15 ml (start volume).
- Incubate 100 min. with 100 rpm at 37 °C.
- Centrifuge 10 min with 3,000 x g at room temperature.
- Aspirate the M-IV medium and remove the last amount of M-IV medium with a pipet.
- Resuspend the bacteria in M-IV medium 10-15 ml (start volume).
- Optional: resuspend into 1 ml for NTHi with lower competence.
- Aliquot 1 ml of competent cells into tubes and add 0.25 ml 80% glycerol and store the bacteria at -80 °C.
- Inoculate 10-15 ml sBHI medium in a 50 ml tube with a few colonies of an overnight sBHI plate and grow to an OD600 = 0.30-0.35 with 200 rpm at 37 °C.
- CTAB genomic DNA isolation protocol
- Inoculate 5 ml sBHI medium in a 50 ml tube with a few colonies of an overnight sBHI plate and grow to an OD600 = 0.30 - 0.35 with 200 rpm at 37 °C.
- Centrifuge 10 min with 3000 x g at room temperature.
- Aspirate the sBHI medium and resuspend the bacteria in 1 ml milli-Q water and transfer to a 2 ml tube.
- Add 70 µl 10% SDS (sodium dodecyl sulphate) and 5 µl 10 mg/ml proteinase K. Mix by inversion, do not vortex. Incubate at 65 °C for at least 10 min.
- Add 100 µl 5 M NaCl, mix and add 100 µl 10 % CTAB (N-Cetyl-N,N,N-trimethylammoniumbromid) in 0.7 M NaCl that was preheated at 65 °C. Vortex until the suspension turns white.
- Incubate at 65 °C for 10 min.
- Extract DNA by adding 500 µl of chloroform\isoamyl alcohol (24:1). Vortex for at least 10 sec.
- Centrifuge 5 min at max in a micro centrifuge.
- Transfer supernatant (~500 µl) to new micro centrifuge tube.
- Add 0.6 volume isopropanol (~300 µl) and mix well.
- Incubate at -20 °C for at least 30 min.
- Centrifuge 10 min at max in microcentrifuge.
- Carefully wash pellet with 500 µl 70% ethanol.
- Centrifuge 5 min at max in microcentrifuge.
- Carefully remove all liquid (if necessary, centrifuge shortly).
- Dry pellet on air (~10 min, pellet becomes white).
- Dissolve pellet in 100 µl Milli-Q water.
- Inoculate 5 ml sBHI medium in a 50 ml tube with a few colonies of an overnight sBHI plate and grow to an OD600 = 0.30 - 0.35 with 200 rpm at 37 °C.
Acknowledgments
This protocol is adapted from a previously published paper: Langereis et al. (2013).
References
- Herriott, R. M., Meyer, E. M. and Vogt, M. (1970). Defined nongrowth media for stage II development of competence in Haemophilus influenzae. J Bacteriol 101(2): 517-524.
- Langereis, J. D., Zomer, A., Stunnenberg, H. G., Burghout, P. and Hermans, P. W. (2013). Nontypeable Haemophilus influenzae carbonic anhydrase is important for environmental and intracellular survival. J Bacteriol 195(12): 2737-2746.
Article Information
Copyright
© 2014 The Authors; exclusive licensee Bio-protocol LLC.
How to cite
Langereis, J. D. (2014). Generation of Non-typeable Haemophilus influenzae Directed Gene Deletion Mutants. Bio-protocol 4(5): e1066. DOI: 10.21769/BioProtoc.1066.
Category
Microbiology > Microbial genetics > Mutagenesis
Molecular Biology > DNA > Mutagenesis
Molecular Biology > DNA > PCR
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.