- Protocols

- Articles and Issues
- For Authors
- About
- Become a Reviewer

Most cited
Most cited in the last five years

Semi-quantitative Determination of Protein Expression Using Immunohistochemistry Staining and Analysis: An Integrated Protocol

Semi-quantitative immunohistochemistry (IHC) is a powerful method for investigating protein expression and localization within tissues that involves using software, such as the freely available Fiji (ImageJ), to conduct deconvolution and downstream analysis. Currently, there is lack of an integrated protocol that includes a detailed procedure on how to measure or compare protein expression. Publications that use semi-quantitative methods to ascertain protein expression often don’t provide enough details in their methods section, which makes it difficult for the reader to reproduce their data. The current protocol provides an example and detailed steps of conducting semi-quantitative analysis of IHC images using Fiji software.
Transcardiac Perfusion of the Mouse for Brain Tissue Dissection and Fixation

Transcardiac perfusion with saline followed by 4% paraformaldehyde (PFA) is widely used to clear blood and preserve brain for immunostaining or in situ hybridization. PFA breaks into formaldehyde in solution, which cross-link protein and DNA molecules to preserve tissue and cell structure. Here we provide a step by step guide for performing this procedure in mouse.

Analysis of Indole-3-acetic Acid (IAA) Production in Klebsiella by LC-MS/MS and the Salkowski Method
Many rhizobacteria isolated from plant rhizosphere produce various phytohormones in the form of secondary metabolites, the most common of which is Indole-3-acetic acid (IAA). Here, we detail analytical protocols of IAA detection and quantification, in vitro and in situ, as recently applied to Klebsiella SGM 81, a rhizobacterium isolated from the rhizosphere of Dianthus caryophyllus (a commercially important flower across the globe). Specifically, we describe a detailed protocol for a colorimetric assay using the Salkowski reagent method, which can be used to screen for the presence of Indole compounds. To further detect and quantify IAA, a highly accurate analytical approach of LC-MS/MS is used. To detect the presence of IAA around the root system of Dianthus caryophyllus, in situ staining of plant roots is done using Salkowski reagent.

Evaluation of Toxicity with Brine Shrimp Assay
The in vivo toxicity of new metallodrugs either as Small Bioactive Molecules (SBAMs) or Conjugates of Metals with Drugs (CoMeDs) or their hydrogels such as with hydroxyethyl-methacrylate (HEMA) (pHEMA@SBAMs or pHEMA@CoMeDs) are evaluated by the brine shrimp assay. Thus individuals of Artemia salina larvae are incubated in saline solutions with SBAMs, CoMeDs, pHEMA@SBAMs or pHEMA@CoMeDs or without for 24 h. The toxicity is then determined in terms of the mortality rate of brine shrimp larvae. Brine shrimp assay is a low cost, safe, no required feeding during the assay, while it requiring only a small amount of the tested agent.

Analysis of Generalized Fibrosis in Mouse Tissue Sections with Masson’s Trichrome Staining
Expansion of fibrous connective tissue and abnormal deposition of extracellular matrix (ECM) are at the basis of many fibrotic diseases. Fibrosis can occur in response to both physiological and pathological cues, including wound healing, tissue remodeling/repair and inflammation. Chronic fibrosis can lead to severe tissue damage, organ failure and death. Assessing the extent of organ fibrosis is crucial for accurate diagnosis of this condition. The use of Masson’s trichrome staining of tissue sections from skeletal muscle is a fast method for detection of morphological alterations indicative of a fibrotic phenotype in this organ. This staining method detects the extent of collagen fibers deposition and, because it employs the combination of three dyes, can also distinguish muscle fibers (red), from collagen (blue) and nuclei (black), simultaneously.
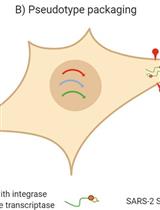
Production, Titration, Neutralisation, Storage and Lyophilisation of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Lentiviral Pseudotypes
This protocol details a rapid and reliable method for the production and titration of high-titre viral pseudotype particles with the SARS-CoV-2 spike protein (and D614G or other variants of concern, VOC) on a lentiviral vector core, and use for neutralisation assays in target cells expressing angiotensin-converting enzyme 2 (ACE2) and transmembrane serine protease 2 (TMPRSS2). It additionally provides detailed instructions on substituting in new spike variants via gene cloning, lyophilisation and storage/shipping considerations for wide deployment potential. Results obtained with this protocol show that SARS-CoV-2 pseudotypes can be produced at equivalent titres to SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) pseudotypes, neutralised by human convalescent plasma and monoclonal antibodies, and stored at a range of laboratory temperatures and lyophilised for distribution and subsequent application.
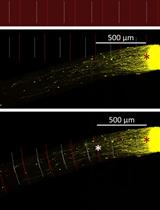
Optic Nerve Crush in Mice to Study Retinal Ganglion Cell Survival and Regeneration
In diseases such as glaucoma, the failure of retinal ganglion cell (RGC) neurons to survive or regenerate their optic nerve axons underlies partial and, in some cases, complete vision loss. Optic nerve crush (ONC) serves as a useful model not only of traumatic optic neuropathy but also of glaucomatous injury, as it similarly induces RGC cell death and degeneration. Intravitreal injection of adeno-associated virus serotype 2 (AAV2) has been shown to specifically and efficiently transduce RGCs in vivo and has thus been proposed as an effective means of gene delivery for the treatment of glaucoma. Indeed, we and others routinely use AAV2 to study the mechanisms that promote neuroprotection and axon regeneration in RGCs following ONC. Herein, we describe a step-by-step protocol to assay RGC survival and regeneration in mice following AAV2-mediated transduction and ONC injury including 1) intravitreal injection of AAV2 viral vectors, 2) optic nerve crush, 3) cholera-toxin B (CTB) labeling of regenerating axons, 4) optic nerve clearing, 5) flat mount retina immunostaining, and 6) quantification of RGC survival and regeneration. In addition to providing all the materials and procedural details necessary to execute this protocol, we highlight its advantages over other similar published approaches and include useful tips to ensure its faithful reproduction in any modern laboratory.
Split-luciferase Complementation Imaging Assay to Study Protein-protein Interactions in Nicotiana benthamiana

The experimental identification of protein-protein interactions (PPIs) is critical to understand protein function. Thus, a plethora of sensitive and versatile approaches have been developed to detect PPIs in vitro or in vivo, such as protein pull-down, yeast two-hybrid (Y2H), co-immunoprecipitation (co-IP), and bimolecular fluorescence complementation (BiFC) assays. The recently established split-luciferase complementation (Split-LUC) imaging assay has several advantages compared to other approaches to detect PPIs in planta: it is a relatively simple and fast method to detect PPIs in vivo; the results are quantitative, with high sensitivity and low background; it measures dynamic PPIs in real-time; and it requires limited experimental materials and instrumentation. In this assay, the amino-terminal and carboxyl-terminal halves of the luciferase enzyme are fused to two proteins of interest (POIs), respectively; the luciferase protein is reconstituted when two POIs interact with each other, giving rise to a measurable activity. Here, we describe a protocol for the Split-LUC imaging assay using a pair of modified gateway-compatible vectors upon Agrobacterium-mediated transient expression in Nicotiana benthamiana. With this setup, we have successfully confirmed a series of interactions among virus-plant proteins, virus-virus proteins, plant-plant proteins, or bacteria-plant proteins in N. benthamiana.

Isolation and Long-term Cultivation of Mouse Alveolar Macrophages
Alveolar macrophages (AM) are tissue-resident macrophages that colonize the lung around birth and can self-maintain long-term in an adult organism without contribution of monocytes. AM are located in the pulmonary alveoli and can be harvested by washing the lungs using the method of bronchoalveolar lavage (BAL). Here, we compared different conditions of BAL to obtain high yields of murine AM for in vitro culture and expansion of AM. In addition, we describe specific culture conditions, under which AM proliferate long-term in liquid culture in the presence of granulocyte-macrophage colony-stimulating factor. This method can be used to obtain large numbers of AM for in vivo transplantation or for in vitro experiments with primary mouse macrophages.
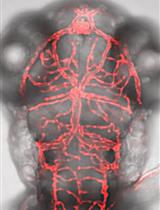
GeneWeld: Efficient Targeted Integration Directed by Short Homology in Zebrafish
Efficient precision genome engineering requires high frequency and specificity of integration at the genomic target site. Multiple design strategies for zebrafish gene targeting have previously been reported with widely varying frequencies for germline recovery of integration alleles. The GeneWeld protocol and pGTag (plasmids for Gene Tagging) vector series provide a set of resources to streamline precision gene targeting in zebrafish. Our approach uses short homology of 24-48 bp to drive targeted integration of DNA reporter cassettes by homology-mediated end joining (HMEJ) at a CRISPR/Cas induced DNA double-strand break. The pGTag vectors contain reporters flanked by a universal CRISPR sgRNA sequence to liberate the targeting cassette in vivo and expose homology arms for homology-driven integration. Germline transmission rates for precision-targeted integration alleles range 22-100%. Our system provides a streamlined, straightforward, and cost-effective approach for high-efficiency gene targeting applications in zebrafish.Graphic Abstract:GeneWeld method for CRISPR/Cas9 targeted integration.
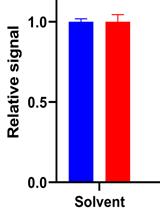
A Simple Microplate Assay for Reactive Oxygen Species Generation and Rapid Cellular Protein Normalization
2',7'-dichlorofluorescein (DCF) and derivatives are commonly used as fluorescent indicators of a broad spectrum of reactive oxygen species (ROS) generation in cell-based assays. However, there are numerous challenges inherent to the utilization of DCF probes for intracellular microscopic analysis, including bleaching and probe efflux. Plate spectroscopy is comparatively simple and scalable compared to microscopy or flow cytometry-based acquisition, however is often subject to artefacts, including those introduced by thermal gradients and normalization methods. In this protocol we demonstrate a simple and sensitive plate spectrometry-based protocol utilizing the probes H2DCFDA and sulforhodamine B. The truncated sulforhodamine B assay (SRB) for cellular protein allows for a stable endpoint measurement of total cell population while also preserving morphology, can be combined or run in parallel with any other assay for normalization of readout to cell mass, and complemented by microscopic scoring of cell number and nuclear count. The oxidative stress and normalisation methods may enhance fields of research investigating cell differentiation, stress, or toxicity.Graphical abstract

Rapid Lipid Quantification in Caenorhabditis elegans by Oil Red O and Nile Red Staining
The ability to stain lipid stores in vivo allows for the facile assessment of metabolic status in individuals of a population following genetic and environmental manipulation or pharmacological treatment. In the animal model Caenorhabditis elegans, lipids are stored in and mobilized from intracellular lipid droplets in the intestinal and hypodermal tissues. The abundance, size, and distribution of these lipids can be readily assessed by two staining methods for neutral lipids: Oil Red O (ORO) and Nile Red (NR). ORO and NR can be used to quantitatively measure lipid droplet abundance, while ORO can also define tissue distribution and lipid droplet size. C. elegans are a useful animal model in studying pathways relating to aging, fat storage, and metabolism, as their transparent nature allows for easy microscopic assessment of lipid droplets. This is done by fixation and permeabilization, staining with NR or ORO, image capture on a microscope, and computational identification and quantification of lipid droplets in individuals within a cohort. To ensure reproducibility in lipid measurements, we provide a detailed protocol to measure intracellular lipid dynamics in C. elegans.Graphic abstract: Flow chart depicting the preparation of C. elegans for fat staining protocols.
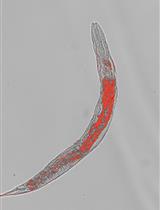
Oil Red O Staining for Lipid Content in Caenorhabditis elegans
The nematode Caenorhabditis elegans has emerged as a popular model system for studying the regulation of lipid metabolism. Therefore, it is critical to develop a method for determining fat storage in individual worms. Oil Red O (ORO) staining has been validated as an accurate assessment for major fat storage in C. elegans. Here, we describe an optimized protocol for ORO staining of C. elegans and provide detailed instructions for quantifying the intensity of ORO signal in images acquired by light microscopy.
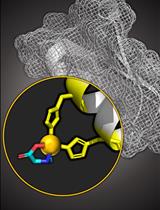
Copper Based Site-directed Spin Labeling of Proteins for Use in Pulsed and Continuous Wave EPR Spectroscopy
Site-directed spin labeling in conjunction with electron paramagnetic resonance (EPR) is an attractive approach to measure residue specific dynamics and point-to-point distance distributions in a biomolecule. Here, we focus on the labeling of proteins with a Cu(II)-nitrilotriacetic acid (NTA) complex, by exploiting two strategically placed histidine residues (called the dHis motif). This labeling strategy has emerged as a means to overcome key limitations of many spin labels. Through utilizing the dHis motif, Cu(II)NTA rigidly binds to a protein without depending on cysteine residues. This protocol outlines three major points: the synthesis of the Cu(II)NTA complex; the measurement of continuous wave and pulsed EPR spectra, to verify a successful synthesis, as well as successful protein labeling; and utilizing Cu(II)NTA labeled proteins, to measure distance constraints and backbone dynamics. In doing so, EPR measurements are less influenced by sidechain motion, which influences the breadth of the measured distance distributions between two spins, as well as the measured residue-specific dynamics. More broadly, such EPR-based distance measurements provide unique structural constraints for integrative structural biophysics and complement traditional biophysical techniques, such as NMR, cryo-EM, FRET, and crystallography.Graphic abstract: Monitoring the success of Cu(II)NTA labeling.
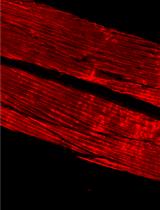
Phalloidin Staining of Actin Filaments for Visualization of Muscle Fibers in Caenorhabditis elegans
Advances in C. elegans research have allowed scientists to recapitulate different human disorders, from neurodegenerative diseases to muscle dysfunction, in these nematodes. Concomitantly, the interest in visualizing organs affected by these conditions has grown, leading to the establishment of different antibody- and dye-based staining protocols to verify tissue morphology. In particular, the quality of muscle tissue has been largely used in nematodes as a readout for fitness and healthspan. Phalloidin derivatives, which are commonly used to stain actin filaments in cells and tissues, have been implemented in the context of C. elegans research for visualization of muscle fibers. However, the majority of the phalloidin-based protocols depend on fixation steps using harmful compounds, preparation of specific buffers, and large amounts of worms. Herein, we implemented a safer and more flexible experimental procedure to stain actin filaments in C. elegans using phalloidin-based dyes. Lyophilization of the worms followed by their acetone permeabilization allows bypassing the fixation process while also providing the opportunity to suspend the experiment at different steps. Moreover, by using conventional buffers throughout our protocol, we avoid the additional preparation of solutions. Finally, our protocol requires a limited number of worms, making it suitable for slow-growing C. elegans strains. Overall, this protocol provides an efficient, fast, and safer method to stain actin filaments and visualize muscle fibers in C. elegans.Graphic abstract:Schematic overview of phalloidin staining in C. elegans for assessing muscle fiber morphology.
Optimized Recombinant Production of Secreted Proteins Using Human Embryonic Kidney (HEK293) Cells Grown in Suspension
Recombinant proteins are an essential milestone for a plethora of different applications ranging from pharmaceutical to clinical, and mammalian cell lines are among the currently preferred systems to obtain large amounts of proteins of interest due to their high level of post-translational modification and manageable large-scale production. In this regard, human embryonic kidney 293 (HEK293) cells constitute one of the main standard lab-scale mammalian hosts for recombinant protein production since these cells are relatively easy to handle, scale-up, and transfect. Here, we present a detailed protocol for the cost-effective, reproducible, and scalable implementation of HEK293 cell cultures in suspension (suitable for commercially available HEK293 cells, HEK293-F) for high-quantity recombinant production of secreted soluble multi-domain proteins. In addition, the protocol is optimized for a Monday-to-Friday maintenance schedule, thus simplifying and streamlining the work of operators responsible for cell culture maintenance.Graphic abstract:Schematic overview of the workflow described in this protocol
A Phenotypic Screen for the Liver Stages of Plasmodium vivax
Control of malaria caused by Plasmodium vivax can be improved by the discovery and development of novel drugs against the parasite’s liver stage, which includes relapse-causing hypnozoites. Several recent reports describe breakthroughs in the culture of the P. vivax liver stage in 384-well microtiter plates, with the goal of enabling a hypnozoite-focused drug screen. Herein we describe assay details, protocol developments, and different assay formats to interrogate the chemical sensitivity of the P. vivax liver stage in one such medium-throughput platform. The general assay protocol includes seeding of primary human hepatocytes which are infected with P. vivax sporozoites generated from the feeding of Anopheles dirus mosquitoes on patient isolate bloodmeals. This protocol is unique in that, after source drug plates are supplied, all culture-work steps have been optimized to preclude the need for automated liquid handling, thereby allowing the assay to be performed within resource-limited laboratories in malaria-endemic countries. Throughput is enhanced as complex culture methods, such as extracellular matrix overlays, multiple cell types in co-culture, or hepatic spheroids, are excluded as the workflow consists entirely of routine culture methods for adherent cells. Furthermore, installation of a high-content imager at the study site enables assay data to be read and transmitted with minimal logistical delays. Herein we detail distinct assay improvements which increase data quality, provide a means to limit the confounding effect of hepatic metabolism on assay data, and detect activity of compounds with a slow-clearance phenotype.Graphical abstract: Overview of P. vivax liver stage screening assay performed at the Institute Pasteur of Cambodia.

Proteome Integral Solubility Alteration (PISA) Assay in Mammalian Cells for Deep, High-Confidence, and High-Throughput Target Deconvolution
Chemical proteomics focuses on the drug–target–phenotype relationship for target deconvolution and elucidation of the mechanism of action—key and bottleneck in drug development and repurposing. Majorly due to the limits of using chemically modified ligands in affinity-based methods, new, unbiased, proteome-wide, and MS-based chemical proteomics approaches have been developed to perform drug target deconvolution, using full proteome profiling and no chemical modification of the studied ligand. Of note among them, thermal proteome profiling (TPP) aims to identify the target(s) by measuring the difference in melting temperatures between each identified protein in drug-treated versus vehicle-treated samples, with the thermodynamic interpretation of “protein melting” and curve fitting of all quantified proteins, at all temperatures, in each biological replicate. Including TPP, all the other chemical proteomics approaches often fail to provide target deconvolution with sufficient proteome depth, statistical power, throughput, and sustainability, which could hardly fulfill the final purpose of drug development. The proteome integral solubility alteration (PISA) assay provides no thermodynamic interpretation, but a throughput 10–100-fold compared to the other proteomics methods, high sustainability, much lower time of analysis and sample amount requirements, high confidence in results, maximal proteome coverage (~10,000 protein IDs), and up to five drugs / test molecules in one assay, with at least biological triplicates of each treatment. Each drug-treated or vehicle-treated sample is split into many fractions and exposed to a gradient of heat as solubility perturbing agent before being recomposed into one sample; each soluble fraction is isolated, then deep and quantitative proteomics is applied across all samples. The proteins interacting with the tested molecules (targets and off-targets), the activated mechanistic factors, or proteins modified during the treatment show reproducible changes in their soluble amount compared to vehicle-treated controls. As of today, the maximal multiplexing capability is 18 biological samples per PISA assay, which enables statistical robustness and flexible experimental design accommodation for fuller target deconvolution, including integration of orthogonal chemical proteomics methods in one PISA assay. Living cells for studying target engagement in vivo or, alternatively, protein extracts to identify in vitro ligand-interacting proteins can be studied, and the minimal need in sample amount unlocks target deconvolution using primary cells and their derived cultures.Graphical abstract:
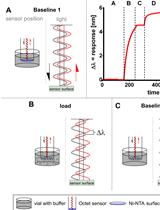
Kinetic Analysis of a Protein-protein Complex to Determine its Dissociation Constant (KD) and the Effective Concentration (EC50) of an Interplaying Effector Molecule Using Bio-layer Interferometry
Biolayer interferometry (BLI) is an emerging analytical tool that allows the study of protein complexes in real time to determine protein complex kinetic parameters. This article describes a protocol to determine the KD of a protein complex using a 6×His tagged fusion protein as bait immobilized on the NTA sensor chip of the FortéBio® Octet K2 System (Sartorius). We also describe how to determine the half maximal effective concentration (EC50, also known as IC50 for inhibiting effectors) of a metabolite. The complete protocol allows the determination of protein complex KD and small molecular effector EC50 within 8 h, measured in triplicates.Graphic abstract:Principle of the Biolayer interferometry measurement. (Middle, top) Exemplary result of the BLI measurement using Octet® (Raw Data). Wavelength shift (Δλ) against time. (A) Baseline 1. Baseline measurement. When the sensor is equilibrated in the kinetics buffer. The light is reflected with no difference. (B) Load. The his-tagged proteins (ligand) are loaded onto the sensor surface. The light is reflected with a shift of the wavelength. (C) Baseline 2. The loaded sensor is equilibrated in the kinetics buffer. No further wavelength shift appears. (D) Association. The loaded sensor is dipped into the analyte solution. The analyte binds to the immobilized ligand along with an increased wavelength shift. (E) Dissociation. Afterward, the sensor is dipped again into the kinetics buffer without the analyte. Some analyte molecules dissociate. The wavelength shift decreases. (Subfigures A-E) The left side shows the position of the sensor during the measurement seen in the representative BLI measurement, marked with the figure label. The right side shows the light path in the sensor. Black waves represent the light emitted to the sensor surface. The red waves show the light reflected from the sensor surface back to the detector.
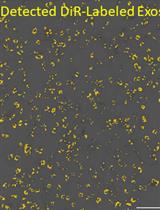
Fluorescent Labeling of Small Extracellular Vesicles (EVs) Isolated from Conditioned Media
Extracellular vesicles (EVs), such as exosomes, are produced by all known eukaryotic cells, and constitute essential means of intercellular communication. Recent studies have unraveled the important roles of EVs in migrating to specific sites and cells. Functional studies of EVs using in vivo and in vitro systems require tracking these organelles using fluorescent dyes or, alternatively, transfected and fluorescent-tagged proteins, located either intravesicularly or anchored to the EV bilayer membrane. Due to design simplicity, the fluorescent dye might be a preferred method if the cells are difficult to modify by transfection or when the genetic alteration of the mother cells is not desired. This protocol describes techniques to label cultured cell-derived EVs, using lipophilic DiR [DiIC18(7) (1,1'-Dioctadecyl-3,3,3',3'-Tetramethylindotricarbocyanine Iodide)] fluorophore. This technique can be used to study the cellular uptake and intracellular localization of EVs, and their biodistribution in vivo, which are crucial evaluations of any isolated EVs.

Protocol Collections
Comprehensive collections of detailed, peer-reviewed protocols focusing on specific topics





