
- Home
- Protocols

-

Cortical excitatory neuron differentiation
Last updated date: Mar 29, 2021 Views: 1518 Forks: 0
Human Excitatory Cortical NGN2 Neuron Differentiation Protocol
*This protocol has been adapted from Zhang et al. 2013. It has two major parts: neural induction via overexpression of NGN2 and dual SMAD & Wnt inhibition and neuronal maturation. Plating density is to be titrated for each cell line as different lines have different viability.
1. Maintenance of ES/iPS cells:
Stem cells are grown on matrigel (Fisher Scientific #BD354277) coated TC dishes/plates and fed daily with mTeSR media (StemCell Technologies #05850). Cultures are passaged as required by accutase (Gibco, A11105) and replated in the presence of 10uM rock inhibitor (1:1000 of 1mM Stock) Y27632 (DNSK International, #129830-38-2) only for the first 24hrs.
2. Schematic
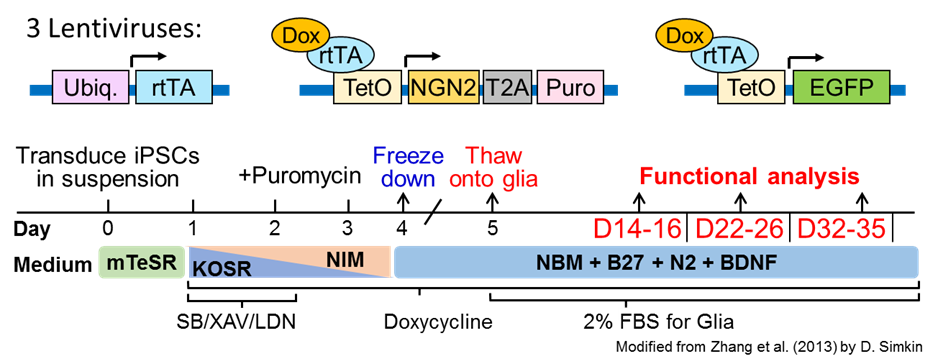
3. Differentiation protocol (example with 2 cell lines)
D0: - Prepare 2x 6-well plates (for differentiation) and 2x 10cm (to passage iPSCs normally) plates by coating all wells with marigel (30 min in hood then 30-60min in incubator). Prep the matrigel coated plates by washing with PBS and then adding 600 ul/well of 6-well or 10mL/ 10cm plate of mTeSR+Rock. Place plates in incubator till you are ready to plate.
- Mix the 3 lentiviruses together in one tube. For 12 wells add 2.4 mL (+ a drop or 2 for pipetting errors) of mTeSR + Rock (~200 µL/well) to a 15 mL conical tube and keep on ice. Add the 3 lentiviruses to this tube. Mix by inverting tube a few times and put back on ice for a few minutes and then split the lentivirus media into 2 separate tubes ~1245 µL each ((2.4 mL + lenti ~90 uL)/2). Let tubes sit on ice until you are ready to mix with cells.
| Lenti viruses (concentrated from NU lenti core): | Quantity per 1 million cells/1 well of 6-well (µL) | For 12 wells of 6-well or 12 million cells multiply by 12 (µL) |
FUW-M2rtTA Addgene #20342 | 4.1 µL | 49.2 µL |
| TetO-Ngn2-Puro Addgene #52047 | 2.9 µL | 34.8 µL |
TetO-EGFP Addgene #30130 | .6 µL | 7.2 µL |
- Passage iPSC (from almost confluent 10cm plate **) with Accutase and resuspend in (3-5 ml) mTeSR+ Rock inhibitor. Count cells (take average of 3 separate counts) and then transfer 0.95–1.1 million cells/well (depending on their rate of growth) to the virus tube. So for 6 wells per line transfer 6 to 6.6 million cells into the viral mixture tube and mix gently by pipetting up and down and then inverting a few times. You should have ~1245uL of virus plus whatever volume of cells you added in your tube which you will later split up to plate into 6 wells.
- Incubate cells/virus mixture in hood at room temp for 4-5 min (with lids slightly unscrewed for oxygen). Mix by inverting a few times half way through your 4-5 min wait (with lids closed!!). After closing the tube tap it a few times on the table to make sure liquid doesn’t get stuck in the cap.
- Calculate volume for 950K-1.1 million cells plus 200 µL (virus mixture). Then pipette cells/lenti mixture to wells. The aim is to have 60-70% confluence by the next day.
** Note: ALL new lenti batches must be titrated to know how much to use. The volumes indicated here are based on NU lenti core facility lenti prep titers.
** Note: (co-infection with TetO-GFP and rtTA but not TetO-Ngn2-Puro is typically used as a control). Virus amount varies with each prep and should be tested prior to scaling up. For a titer of 109, use 0.1ul of virus per 50,000 cells (always use more rtta though).* titer each virus batch before use!!!
** Note: In the case that stable iPSC lines have already been infected with the lenti viruses are used, just plate iPSCs without viral transduction and start D1 the following day.
** Note: 3 wells of a 6-well plate of stem cells will generate 5-8 million NGN2 neurons
D1: Feed (2 mls/well) with KOSR media + SB/XAV/LDN. Add Doxycyclin (1:6667; 1.5x) to induce NGN2 and GFP expression. Keep Dox in all media from this day onwards.
** Keep one well/line as stem cells and feed mTeSR until they are ready to passage to expand. Freeze ~1 million NGN2 transduced iPSCs and passage the rest onto 1 or 2 10cm plates. When these plates are almost confluent passage them again onto 2-4 10cm plates at 6 -7 million cells per plate (and make a freeze). The next day (D1) feed with D1 media (KOSR + SB/XAV/LDN plus Doxycyclin (1:6667; 1.5x)).
D2: Feed with 1:1 ratio of (KOSR + SB/XAV/LDN): (NIM) and add Dox (1:6667; 1.5x). Start selection by adding Puromycin (1:5000) to select for infected cells.
**Note: NIM is very sensitive and turns pink quickly which will affect differentiation qualityso make it from newly opened bottles of DMEM/F12, make small volumes andonly keep it for 2-3 days at 4⁰C.
D3: Feed with NIM media + Dox (1:6667; 1.5x), add Puromycin (1:5000).
D4: Freeze down: Use accutase 4-5min to dissociate cells, wash and spin down in neurobasal base media + puromycin (1:5000). Resuspend cells in NBM +Dox (1:6667; 1.5x), +BDNF and Rock inhibitor. Count cells (take average of 3 counts) and freeze down (in freezing container in -80C freezer) at 1-2 million cells per vial in 10% DMSO/Hyclone FBS. Move cells to liquid nitrogen within 2 days.
**Note: Freezes with higher density of neuron freezes tend to have higher after thaw viability but might be a waste of cells depending on experiment. Instead you can reduce the volume in which you freeze in (e.g. instead of 1ml freezes freeze in 500uL/vial).
For freezing neurons from 10cm plates make freezes of ~8-12 Million neurons per vial and whatever is left over so that you have relatively even numbers of high concentration vials. The goal is to have about 50-60 million neurons when you freeze down which equals about 30- 35 million at thaw. It's fine if you have less but it’s better to have multiple freezes of the same neurons for Q-state.
Viruses (viruses need to be titrated first since every batch may be at different concentration):
- TetO-Ngn2-Puro; addgene: #52047: use 2.8-80 uL/ 1 Million cells
- FUW-M2rtTA (Ubiq promotor); addgene #20342: use 3.8-80 uL/ 1 Million cells
- TetO-FUW-EGFP; addgene #30130: use .5-40 uL/ 1 Million cells
- Or TetO-FUW-RFP; Made by DSi: use .5-40 uL/ 1 Million cells
** Note: Only use high titer (>109) virus for infections
Thawing and plating NGN2 neurons
| Plate type (1 well) | # of Glia | # of Neurons | Purpose |
| 12mm coverslip in 24-well plate | 90K drop | 35-40K | ephys/imaging |
| 1w of 6-well | 200-250K | 450K-500K | qPCR/Western/sorting |
| 1w of 12-well MEA | 40-50K drop | 30-35K | MEA recording |
| 1w of 48-well MEA | 40-50K drop | 30-35K | MEA recording |
| 1w of 96-well Vala plate | 25-30K | 15K | imaging |
- Coat plates/coverslips with PDL overnight in 4°C. Next day wash plates x2 water and x1 with PBS and let dry completely (10-15 min in hood; make sure coverslip in the center of the well).
- Coat plates with 20 uM laminin diluted in PBS for at least 2 hour in incubator before plating mouse glia. When ready to plate glia aspirate laminin but DO NOT WASH IT, and let well dry a little bit while glia is spinning down (~2-4min).
- Plate mouse glia (P2) in glia media to make monolayer at least 4 days before neurons.
** Note: When plating glia, plate in volume of 50-70µl or 20µL drop of media, in center of coverslip or MEA well, respectively. Incubate 5-10 minutes for cells to adhere, then add 1 mL glia media on top.
D5: Thaw out frozen neuronsinto neurobasal +puro (1:5000)/ spin down and resuspend in NBM media + BDNF (1:10000), Dox (1:6667; 1.5x), 2% FBS and Rock inhibitor. Count and plate onto glia monolayer. Next day do half media change to NBM media + BDNF(1:10000), Dox (1:6667; 1.5x) + 2% FBS.
** Note: Usually you will get a little over half of the neurons frozen surviving the thaw.
D7-36: Continue to feed (half change) every 2-3 days (Mon, Wed, Fri) with NBM base media + BDNF/Dox (1:10000; 1x) + 2% FBS.
** Note: On Fridays make sure cells get extra media for the 2 day weekendand if neurons start to look clumpy (cells that didn’t get enough virus still dividing or glia disappearing) add laminin to media.
*** If there are too many progenitor or dividing cells taking over you can pulse Ara-C (1:10000) so Day 7 add Ara-C (1:5000 since doing half change) and then continue half-change feedings with normal media from then on (NBM + BDNF/Dox (1:10000; 1x) + 2% FBS).
Reagents: *It is important to stick to the same lot numbers for entire experiment because there may be differences between lot numbers of reagents.
| Reagent | Vendor | Cat no. | [Stock] | [Final] | Dilution |
| LDN-193189 | DNSK International | 1062368-24-4 | 1 mM | 100 nM | 1:10000 |
| SB431542 | Tocris | 1254 | 10 mM | 10 uM | 1:1000 |
| XAV939 | Stemgent | 04-00046 | 10 mM | 2 uM | 1:5000 |
| Doxycycline | Sigma Aldrich | D9891-1G | 20 mg/mL | 2 ug/mL | 1:10000 |
| Puromycin | Sigma-Aldrich | P8833 | 10 mg/mL | 2 ug/mL | 1:5000 |
| BDNF | R&D Systems | 248-BD | 100 ug/mL | 10 ng/mL | 1:10000 |
| Laminin | Life Technologies | 23017-015 | 1 mg/mL | 20 ug/mL | 1:50 |
| Heparin Sulfate | Sigma Aldrich | H3149 | 50 mg/mL | 2 ug/mL | 1:25000 |
| BME (diluted) | Gibco™ (thermofisher) | 21985023 | 55 mM | 55ul | 1:1000 |
| ***Ara-C | Sigma Aldrich | C1768 | 40mM | 4uM | 1:10000 |
Media Recipes:
KOSR (Knockout Serum Replacement Medium) scaled down to 50 mL
41.5 mL Knockout DMEM (Life Technologies # 10829-018)
7.5 mL Knockout Replacement Serum KSR (15%) (Life Technologies # 10828028)
500 µL MEM non-essential amino acids (NEAA; Life Technologies # 10370088)
500 µL Glutamax (Life Technologies # 35050061)
50 µLbeta-mercaptoethanol (diluted stock)
Add BME after sterile filtering, store for <2 weeks 4°C, light protected
NIM (Neural Induction Medium) scaled down to 50 mL (NIM tends to turn pink very fast)
48.1 mL DMEM/F12 + L-glutamine (Life Technologies #11320-033)
500 µL Glutamax
500 µL NEAA
400 µL 20% D-glucose in H2O (Sigma Aldrich # G8769-100ML 45% D-glucose)
500 µL N2 supplement (Life Technologies # 17502048)
2 µL Heparin Sulfate (HS; 2 ug/ml final conc.)
Add N2 and Heparin Sulfate after sterile filtering, store for <1 week 4°C, light protected
NBM (NeuroBasal Medium): 500 mL
475 mL Neurobasal+ L-glutamine (Life Technologies # 21103049)
5 mL Glutamax
5 mL NEAA
5 mL N2 supplement
10 mL B27 supplement (Life Technologies # 17504044)
Add N2 and B27 after sterile filtering, store for <2 weeks 4°C, light protected
Glia Media: 500 mL
445 mL DMEM (cat #: 15013CV)
5 mL Glutamax (cat #: 35050061)
50 mL HI Horse Serum (cat#: 26050-088)
Filter store at 4°C for <3 weeks or if it turns pink. Change glia media at least x1 a week.
- Simkin, D(2021). Cortical excitatory neuron differentiation. Bio-protocol Preprint. bio-protocol.org/prep981.
- Simkin, D., Marshall, K. A., Vanoye, C. G., Desai, R. R., Bustos, B. I., Piyevsky, B. N., Ortega, J. A., Forrest, M., Robertson, G. L., Penzes, P., Laux, L. C., Lubbe, S. J., Millichap, J. J., George, A. L. and Kiskinis, E.(2021). Dyshomeostatic modulation of Ca2+-activated K+ channels in a human neuronal model of KCNQ2 encephalopathy. eLife. DOI: 10.7554/eLife.64434
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.