
- Home
- Protocols

-

C2C12 culture and generation of Gene-modified C2C12 cells
Last updated date: Dec 4, 2019 Views: 1375 Forks: 0
Generate CRISPR construct:
How to design the primers:
To generate the CRISPR-Cas9 plasmid for the gene you interested, we have to choose a target sequence with high efficiency and low off-target possibility. This website provides these information for the target sequence
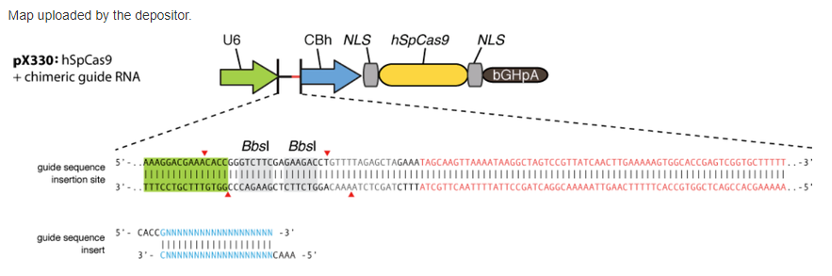
Here, we use pX330 as an example (if you want to generate knockout or knockin cell line, please use pX330-2A-GFP vector, which puromycin in pX330 was replaced by eGFP):
As shown in the figure,
Forward primer : CACCGNNNNNNNNNNNNNNNNNNNN (N is the sequence you selected to target)
Reverse primer: AAACNNNNNNNNNNNNNNNNNNNNC (N is reverse complement to the forward N)
How to generate constructs:
1. Align two oligo DNA (primers) to double strand DNA
Oligo F (10 uM) 9 ul
Oligo R (10 uM) 9 ul
10* T4 ligase buffer 2 ul
20 ul
95℃ 5 min
0.1℃/sec to
16℃ 10 min
2. pX330 is digested by BbsI
H2O 16 ul
10*Buffer 2.1 2 ul
pX330 1 ul
BbsI 1 ul
20 ul 37℃ 1 h
3. Insert the aligned oligo into digested pX330
pX330 digestion (from step 2) 20 ul
aligned oligo (from step 1) 2 ul
10* T4 ligase buffer 2.5 ul
T4 ligase 0.5 ul
25 ul 37℃ 1 h
4. Transform 10 ul of the ligation product into competent cells.
The positive clones can be identified by PCR using U6 forward and oligo Reverse primers.
The positive clones should be confirmed by sequencing results.
Generation of knockout/knockin cell lines:
- Plate the cells in the 10-cm dish (to get enough cells for sorting).
- Transfect the pX330-2A-GFP into cells. (When you sort GFP-positive cells, you may need the non-transfected cells as negative control).
- If you want to generate knockin cells, prepare the donor DNA and transfect the donor DNA together with pX330-2A-GFP. (both plasmid and PCR product could be used as donor. The homology arms can be as short as 100 bp, but longer arms ~500 bp are highly recommended)
- 48 hours after transfection, sort GFP positive cells into 96-well plate by FACS. (one cell per well)
- Each cell may grow up to a knockout cell line. Freeze some when you have enough cells.
Identification of Knockout cells:
- Western-blot is always a good way to identify the knockout clones.
- PCR sequencing is an alternative way to determine gene editing at DNA level.
Design primers ~200 bp up/down stream of the sequence you manipulated.
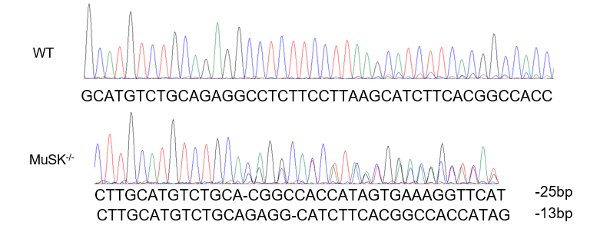
Get the genomic DNA of each cell lines and PCR amplify the targeting DNA. The PCR product will be sequenced by forward or reverse primer you designed. - Once you see double peaks, the CRISPR works. You may determine how the DNA is repaired after double-strand break.
- Mei, L(2019). C2C12 culture and generation of Gene-modified C2C12 cells. Bio-protocol Preprint. bio-protocol.org/prep93.
- Xing, G., Jing, H., Zhang, L., Cao, Y., Li, L., Zhao, K., Dong, Z., Chen, W., Wang, H., Cao, R., Xiong, W. and Mei, L.(2019). A mechanism in agrin signaling revealed by a prevalent Rapsyn mutation in congenital myasthenic syndrome. eLife. DOI: 10.7554/eLife.49180
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.