
- Home
- Protocols

-

Hfq UV cross-linking, ligation and analysis of hybrids (Hfq-CLASH)
Last updated date: Nov 3, 2020 Views: 1103 Forks: 0
CCLASH Protocol for E. coliLASH Protocol for E. coli
Introduction
CLASH protocol was originaly designed by Sander Granneman and later optimised for Staph by Stuart McKellar.
Materials
- 0.45um filters for bacteria
- Lysostaphin 10mg/ml
- RQ1 DNase
- Superase-In
- Zirconia beads 0.1mm
- 1M Tris-HCL pH7.5-7.8
- 1M NaCl
- 10% NP-40
- 2M beta-mercaptoethanol
- TritonX-100
- TritonX-100 20%
- 1M MgCl Sigma
- 5M NaCl
- RocheEDTA-free protease inhibitor
- 0.5M EDTA comersial
- 2.5M Imidazole pH7.5
- GuHCl
- FLAG Magnetic beads
- TEV protease
- NTA beads Quiagen
- Rnase-It
- Pierce spin columns
- TSAP
- rRNasin
- 32P-gamaATP
- T4PNK
- ATP 100mM
- T4 RNA ligase I (NEB)
- 5'adaptor (100uM)
- ATP 10mM
- APP-PE 3'adapter(100uM) -80C
- T4 RNA ligase 2 truncated K227Q
- 50% PEG 8000
- TCA
- glycogen
- acetone
- NuPAGE 4x buffer
- 4-12%Bis-Tris gel
- 20x NuPAGE Running Buffer
- Film
- proteinaseK
- phenol-chloroform-IAA acid
- 3M NaAc pH5.2 commersial
- Absolute ethanol
- 70%Ethanol
- 5mM dNTP
- RT oligo #409 App-PE-RT
- 5xSSIV buffer
- 100mM DTT
- Superscript IV
- RNaseH (NEB)
- RNACleanX beads
- 10xPFU buffer
- P5 primer Fw
- BC primer Rev
- PFU polymerase
- exonuclease I
- 6%TBE gel
- 50bp Ladder
- Glass filters and CoSTARX spin columns
- 1kB Ladder
- 1xTBE buffer
Procedure
Bacterial growth
- Prepare 1x 200 mL of the desired medium.
- Inoculate 1x 200 mL to OD600 = 0.05 or 0.1. Grow until the desired OD600
- Collect liquid nitrogen
Lysis
- Resuspend cells from filter in ~30 mL cold PBS. Transfer to new 50 mL Falcon. Centrifuge at 4000 rpm for 8 minutes to collect.
- Remove SN, resuspend in 2V of TN150 buffer and transfer to an 5mL srewcap tube. Add 60 µL of RQ DNase 1 and 10 µL Superase-In, and incubate at 20°C for 30 mins
- Add 3V (in mLs) of Zirconia beads to the cell pellet. Vortex 1 min, then 1 min on ice. Repeat for a total of five times
- Add 2V cold TN150 anti-peptidase buffer
- Centrifuge the suspension in the Falcon tube for 40 minutes at full speed in the Heraeus centrifuge (4600 g) at 4°C
- Lysate gel sample for WB 20uL
FLAG capture
- Thaw beads on ice
- Cut a pipette tip, and take 37.5 µL of beads (dry) per sample (75 µL slurry). (I used to take separate aliquots for each sample and wash them separately.)
- Wash 3x in TN150, using magnetic tube rack with 1mL
- Upon last wash, remove all SN and resuspend beads in a known volume.
- Add equal amount of beads to each cleared lysate. (I add the lysate to the beads in this step.)
- Incubate beads for 2 hours with rotation at 4°C
- Prepare 10x core buffer as it requires time for the TritonX100 to disolve
NO EDTA TO BE USED HEREAFTER
Reducing, oxidising agents and chelators such as EDTA, DTT and SDS are incompatible with Ni- purification.
TEV digestion
- Flow through gel sample for WB
- Wash beads 3 times in TN1000 for 10 mins at 4°C with rotation with 1mL. This step can also be done for 5' at room temperature with 2mL.
- Rinse beads 3 times with TN150 and remove all buffer in final wash 2mL
- Resuspend beads in 250 µL of TN150. Transfer to new Eppendorf
- Add 10 µL of TEV and incubate at RT for 2 hours
Meanwhile, wash NTA beads in WB I 1mL (50 µl beads per sample, 100 µl of 50% nickel slurry) and prepare future tubes:
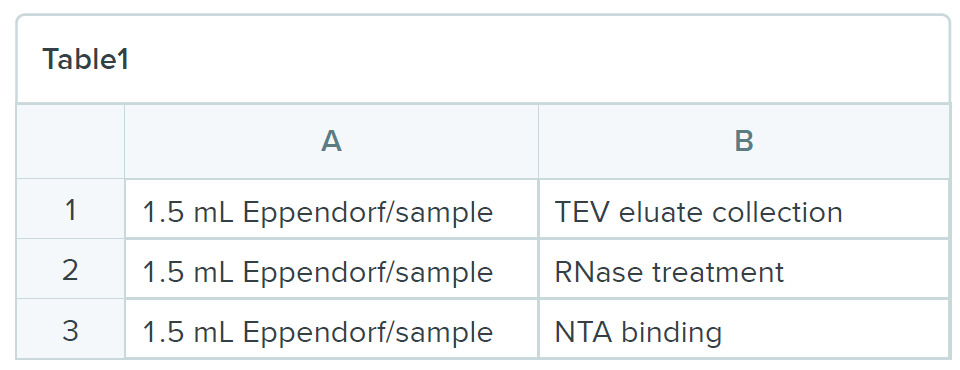
- To NTA tubes, add 0.4 g GuHCl, 3 µL of 2.5M imidazole and 27 µL of 5M NaCl
- Collect TEV eluates and transfer to the TEV eluate collection tube. Add 350 µL of TN150
- TEV elute gel sample (50 µL). I keep the left overs in the TEV elute collection tube.
RNase digestion and nickel purification
Different metal ions can be used such as cobalt and copper. Cobalt is the most specific binder and copper is the strongest. Nikel is between so the best balance between specificity and strength.
- Set incubator to 22°C
- Add 1 µL of RNace-It (1/100 dilution) to the RNase treatment tubes
- Take 550 µl of TEV eluate from sample 1, put onto RNace-It, put into incubator, set timer
- Repeat for remaining samples
- Incubate for exactly 7 minutes. After 7 minutes, start to remove tubes one-by-one
- Transfer 500 µl of the solution to the NTA binding tubes (containing GuHCl, imidazole and NaCl)
- Vortex to dissolve the GuHCl
- Add equal amounts of the previously washed NTA beads and rotate overnight at 4°C
- STOPING POINT
Alkaline phosphatase treatment (3’ dephosphorylation) of precipitated RNAs
- Set incubator to 20°C
- Transfer Ni2+ beads to Pierce spin columns
- To get the remainder of the beads out of the Eppendorf, wash with WB I and transfer to the spin columns
- Wash the beads 2x more with WB I and then 3x with NP-PNK
- Spin beads dry and put on stopper (Always put on and take out the stopper on open cap tube to release first the pressure! )and add 60uL:
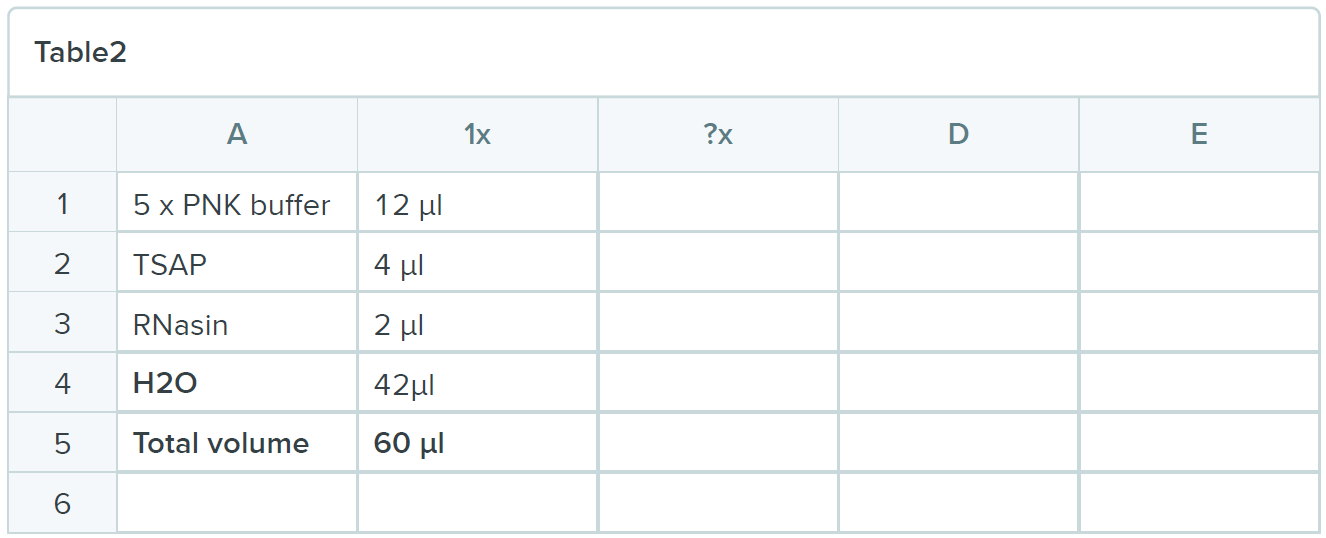
- Incubate for 1 hour
- Wash the beads 1x more with WB I 500uL and then 3x with NP-PNK 500uL
Phosphorylating the 5’ ends of the RNA and radiolabeling
Spin out the remaining buffer and add 80 µl of the following mix:
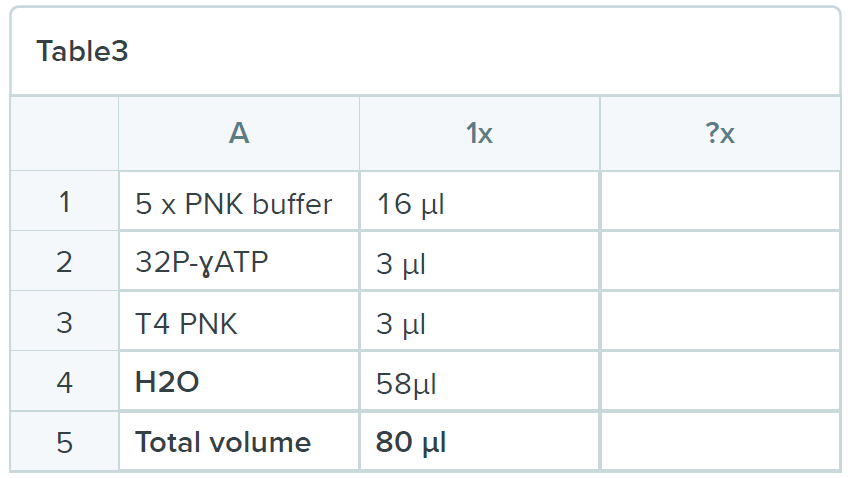
- Incubate the reaction for 100 minutes at 20ºC
- Add 1 µl 100 mM ATP and let the reaction proceed for another 50 minutes. This will make sure that almost all of the 5’ ends have phosphates
- Set up 7 rows of tubes
- Wash beads three times with 500 µl of wash buffer I and three times with 500 µl NP-PNK buffer
- Initial washes very hot: add to 15 mL Falcon and then highly-radiative waste jar
On-bead ligation of the 5’ linker
Spin out the remaining buffer and add 74 µl of the following mix:
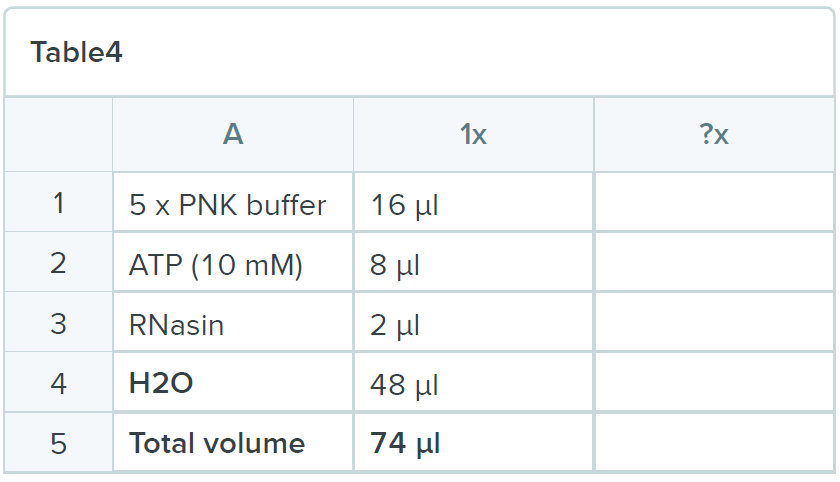
- Add 2 µl of 5’ adapter (100 µM) and 4 µl of T4 RNA ligase I (NEB) to each tube and incubate for 16 hours at 16ºC
- STOPING POINT
On-bead ligation of the App-PE linker to the 3' end of the RNA
- Wash 1x WB I and 3x NP-PNK
- Spin out the remaining buffer and add 60 µl of the following mix:
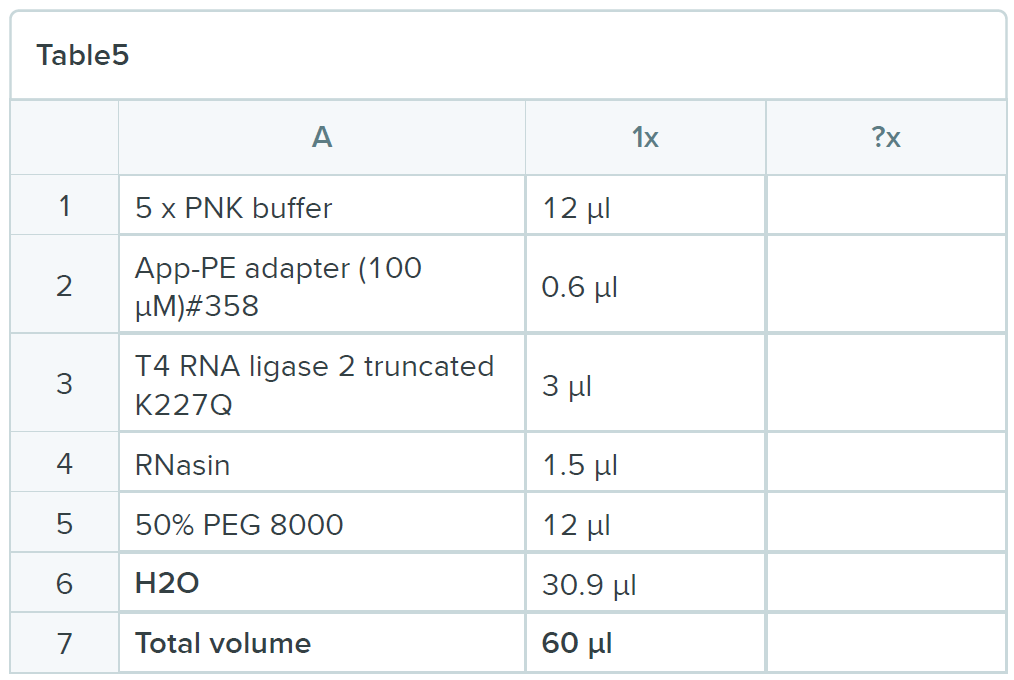
- Incubate the reaction for 6 hours at 25ºC
- Wash beads once with 500 µl of wash buffer I and 3x 500 µL WB II
- Spin out void volume
Elution
- Prepare 2 rows of 1.5 mL Eppendorfs for elution. Add 2 µL of 20 mg/ml glycogen to the second. Cool centrifuge
- Spin out the void volume and elute with elution buffer:
- Add 200 µL
- Wait 2 minutes
- Force elution buffer through column by opening/closing cap
- Transfer column to new tube (containing glycogen)
- Add 200 µL
- Wait 2 minutes
- Force elution buffer through column by opening/closing cap
- Spin out
- Pool by transferring from tube 1 to tube 2
- Add 100 µL TCA to each ~400 µL eluate, vortex well, let sit on ice in fume hood for 20 min, spin at 13.4K, 4°C, 30 min
- CAREFULLY remove supernatant (pellet may be invisible. Remove SN in turns, ensuring no/little radioactivity in SN, and all in pellet. Any liquid with significant radioactivity = return to tube)
- Resuspend pellet in 800µl acetone, spin 13.4K, 4°C, 15 min
- Remove acetone completely, dry pellet for a few minutes (NOT LONGER!) with lid open in the fume hood
- Resuspend in 20 µL 1x NuPAGE buffer
- Potential stop point: freeze LDS samples at -20°C overnight
- Boil samples at 65°C (NOT 95!)
- Load on NuPAGE 1mm 4-12% Bis-Tris gel, run for 1.5 hours at 125V
- Expose a film for ~1.5-3 hours at -80°C
- NOTE: wrap the gel (I keep one of the plastic parts as well) in cling film before putting a film on and freezing it. After developing the film, ensure that you allow the gel to FULLY THAW to room temperature before removing the cling film and cutting it. Otherwise, the gel shrinks and your desired band moves.
- Develop film 3h or over night.
- STOPING POINT
Proteinase K treatment
- Excise protein regions from gel and place into2mL Epppi.Smash into pieces with 1mL tip, leave tip inside the tube, then add 600uL of port K buffer. Try to wash gel pieces stuck to tip off with the buffer.
- Add 8uL of 20mg/ml proteinase K
- Incubate for 2 hours at 55oC with 800-900 rpm shaking.
- Use Mobicol spin tube to harvest SN from gel pieces and transfer to a new tube. Here you mix control and experimental as they have different 5' adaptors. This way you won't overamplify background sequences and overtake the sequencing. It is also possible to pool all your samples and continue with one library.
- Add 1/10 vol 3M NaAc ph5.2, 1vol of bottom phase phenol/chloroform mix
- Vortex 1 min, centrifuge 5-7 min at max.
- Remove top layer, transfer to new tube.
- Add 1vol chloroform, vortex, centrifuge for 5-7 min at max.
- Take off top layer, put in a new tube with 2 uL 20mh/ml glycogen. Add 2.2 vol EtOH 96% ice cold
- Incubate in -80oC for 1 hour
- Spin at 13.4K, 30 min, 4oC. Wash with ice cold 70% EtOH, remove and air dry then dissolve RNA in 20uL water.
Reverse transcription
Reaction mix: resuspend the RNA pellet in the following mix:
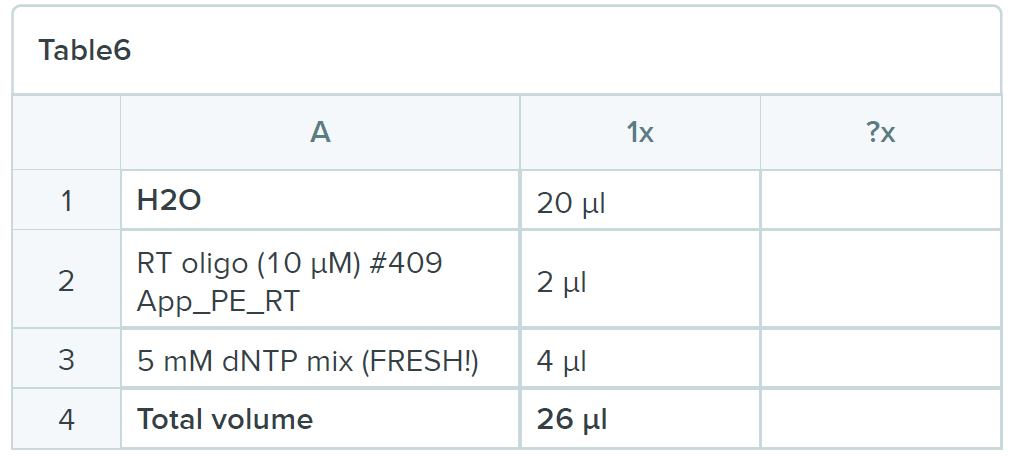
Heat mixture to 85ºC for 3 minutes and snap chill on ice for 5 minutes. Collect the contents by brief centrifugation and add 12 µl of the following mix:
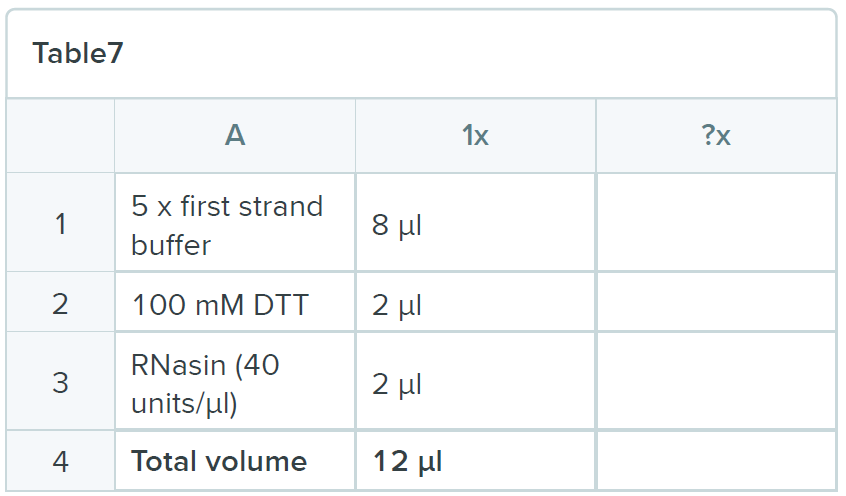
- Incubate the mixture at 50ºC for three minutes and then add 2 µl of Superscript III and incubate the reaction for one hour at 50ºC
- Inactivate Superscipt IV by incubation at 65ºC for 15 minutes
- Transfer the tubes to 37ºC and leave them there for 3 minutes
- Add 2 µl of RNase H (NEB) and incubate for 30 minutes at 37ºC
RNA Clean XP beads
- Isolate the cDNAs using RNA Clean XP beads:
- Transfer the cDNA to PCR tube
- Add 84uL of the beads and pipet to mix well
- Leave for 15min on bench
- Place on the magnet wait 5 min for the solution to clear
- Discard the supernatant
- Wash 2x with ice cold 70%EtOH (I use fresh made one).
- Leave to dry from the last wash <10 min
- Resuspended in 12uL pipet for couple minutes and place on the magnet
- Collect 11uL and place in a new Eppi, store at -20oC.
- STOPING POINT
PCR reaction and gel extraction
- Run PCR
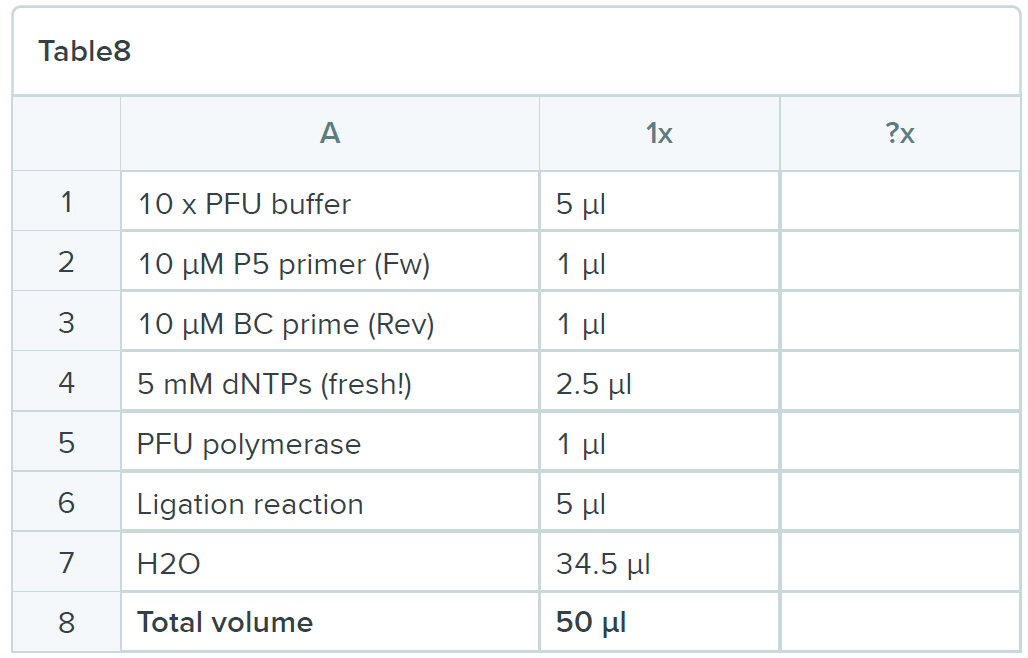
P5-P3 Program:
95 ºC 2 min98 ºC 20 sec
52 ºC 30 sec
72 ºC 1 minute
24 cycles (this needs to be carefully controlled as you don’t want to “over amplify”. Use your negative control also as a negative control in PCR.
72 ºC 5 min
- Add 1 µL of exonuclease I and incubate at 37°C for 60 min
RNA Clean XP beads
- Isolate the cDNAs using RNA Clean XP beads:
- Transfer the PCR product to PCR tube
- Add 100uL of the beads and pipet to mix well
- Leave for 15min on bench
- Place on the magnet wait 5 min for the solution to clear
- Discard the supernatant
- Wash 2x with ice cold 70%EtOH (I use fresh made one).
- Leave to dry from the last wash <10 min
- Resuspended in 12uL pipet for couple minutes and place on the magnet
- Collect 21uL and place in a new Eppi, store at -20°C.
Gel purification
- Run on 6%TBE gel for 1h at 100V with 50bp and 1kB Ladder. Leave spaces in between samples and ladders.
- Stain with Syberdye (3uL in TBE covering the gel in one of WB boxes)
- Place the gel in transparent folder not brake the gel.
Check on Phosphoimager at Tollervey lab
1 laser 1 imageSYBR 400 FITC, wavelength = 473 nm, FLA-5000
200 resolution
Select area
Print with inverted colours, 100X size
- Cut the band
- Smash it with 1mL tip in 1.5mL Eppendorf
- Add 400uL of water and incubate at 37°C for 1h and 1100rpm
- Freeze on dry ice for 10min
- Place back at 37oC for 1h and 1100rpm
- Spin briefly place the liquid in 2 glass filters in a Costar filter collumn
- Keep the supernatant and add 1uL of glycogen, 40uL of 3M NaAc pH5.2 and 1mL of 96%EtOH
- Incubate for 30min at -800C/or overnight at -20°C
- Spin at max for 15 min 4oC
- Discard supernatant and wash with 500 uL 70% EtOH
- Spin 5 min max
- Resuspend in desired volume (10uL)
- Measure concentration on Qbit HS DNA and store at -20oC.
BUFFERS
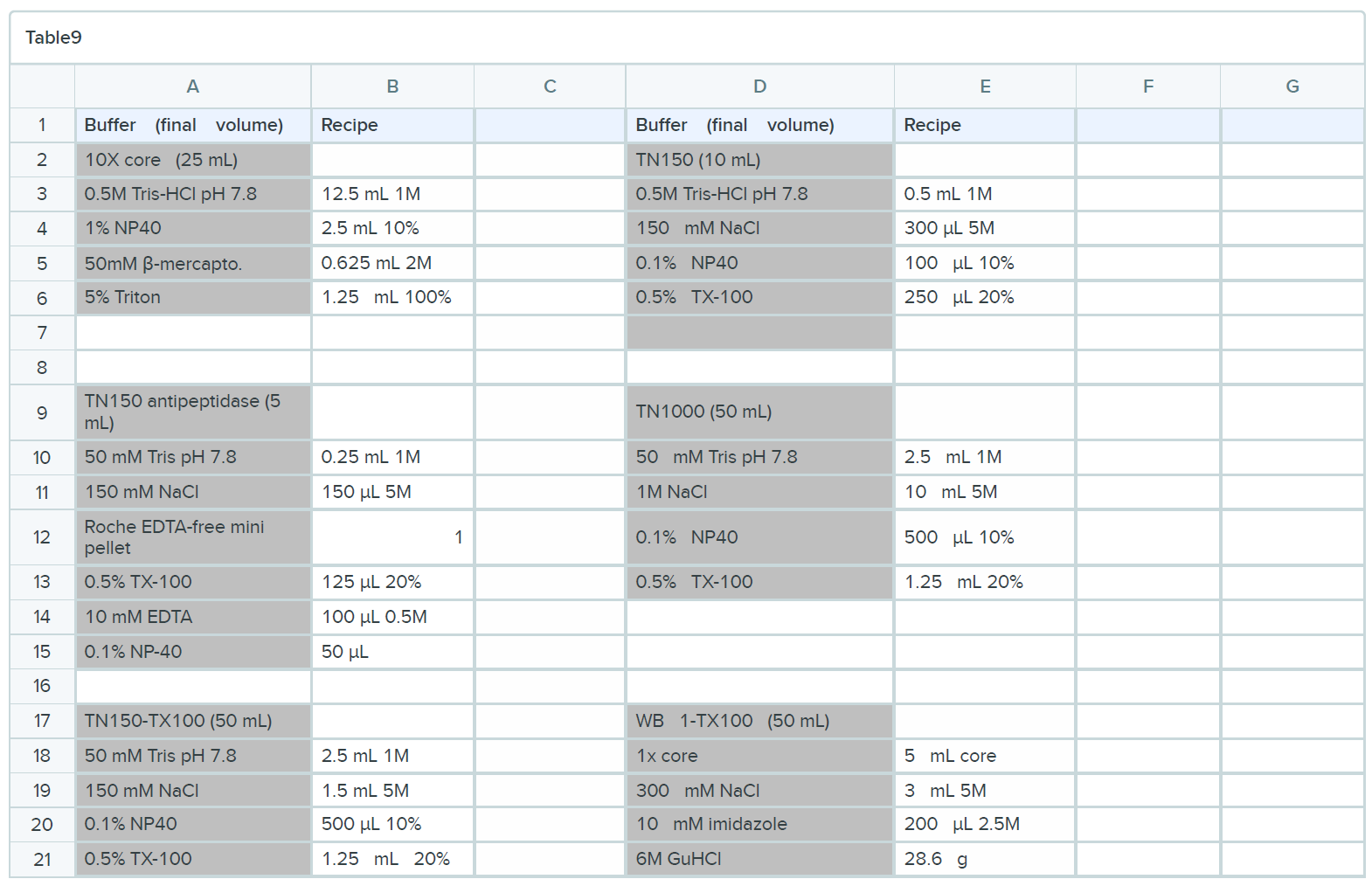
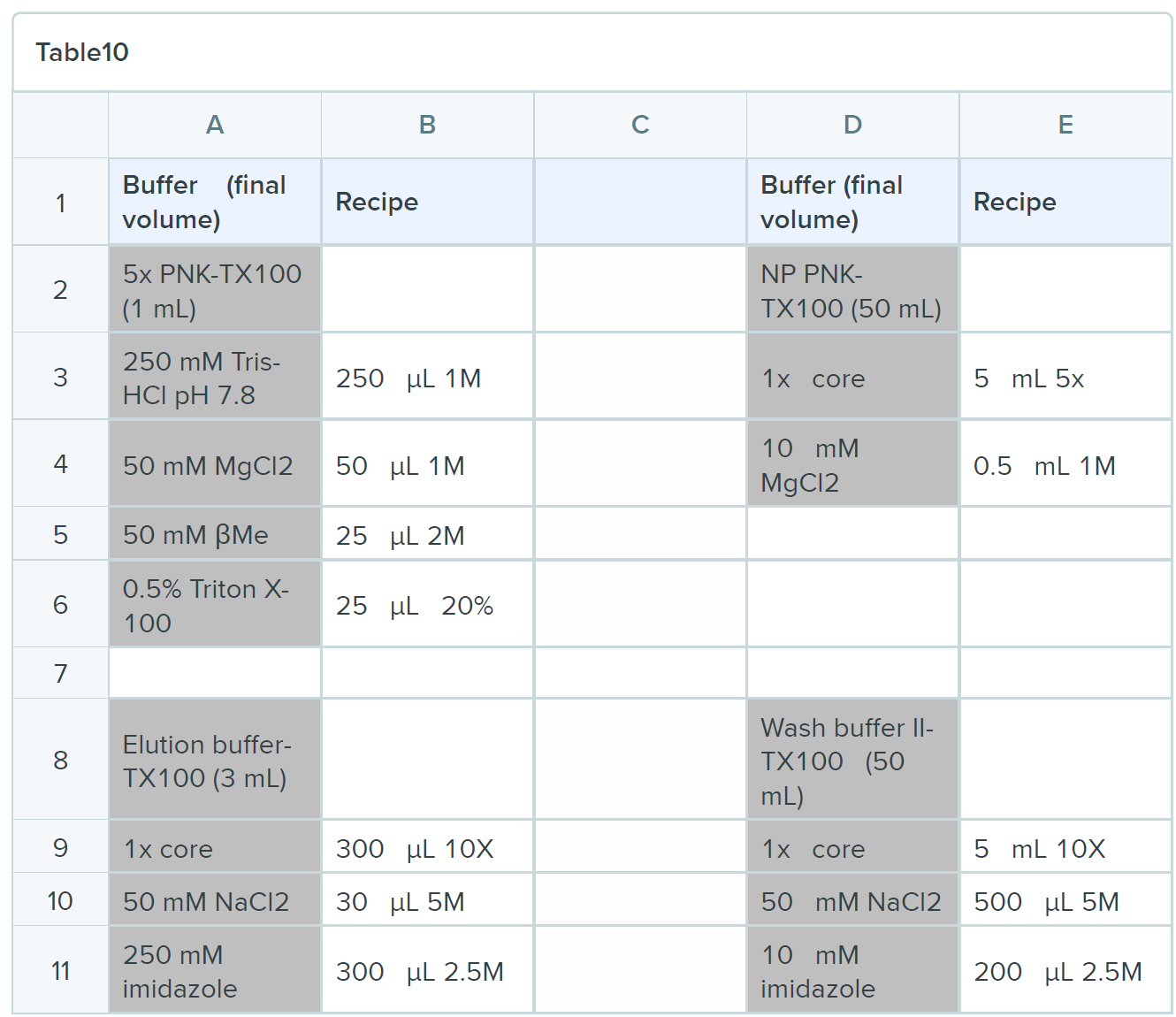
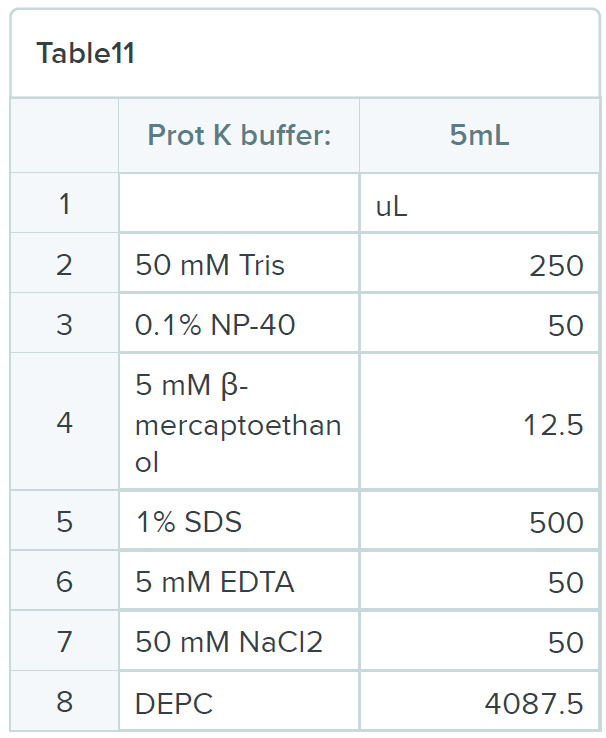
Oligonucleotides
5’ adapters
BARCODE SET A
BARCODE SET A
|
L5Aa 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrUrArArGrCrN-OH-3' |
L5Ab 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrArUrUrArGrCrN-OH-3' |
L5Ac 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrGrCrGrCrArGrCrN-OH-3' |
L5Ad 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrCrGrCrUrUrArGrCrN-OH-3' |
BARCODE SET B |
L5Ba 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrArGrArGrCrN-OH-3' |
L5Bb 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrGrUrGrArGrCrN-OH-3' |
L5Bc 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrCrArCrUrArGrCrN-OH-3' |
L5Bd 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrUrCrUrCrUrArGrCrN-OH-3' |
BARCODE SET C |
L5Ca 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrCrUrArGrCrN-OH-3' |
L5Cb 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrUrGrGrArGrCrN-OH-3' |
L5Cc 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrArCrUrCrArGrCrN-OH-3' |
L5Cd 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrGrArCrUrUrArGrCrN-OH-3' |
BARCODE SET D |
L5Da 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrCrGrUrGrArUrN-OH-3' |
L5Db 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrGrCrArCrUrArN-OH-3' |
L5Dc 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrUrArGrUrGrCrN-OH-3' |
L5Dd 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrArUrCrArCrGrN-OH-3' |
BARCODE SET E |
L5Ea 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrCrArCrUrGrUrN-OH-3' |
L5Eb 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrGrUrGrArCrArN-OH-3' |
L5Ec 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrUrGrUrCrArCrN-OH-3' |
L5Ed 5'-invddT-ACACrGrArCrGrCrUrCrUrUrCrCrGrArUrCrUrNrNrNrArCrArGrUrGrN-OH-3' |
3’ adapters:
App_PE: 5’App-NAGATCGGAAGAGCACACGTCTG-ddC 3’
RT oligonucleotides:
5’-CAGACGTGTGCTCTTCCGATCT-3’
PCR oligonucleotides:
P5 (Forward)
5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCT-3’
P3 (Reverse; BC1-BC6)
BC1 5’-CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3’ BC2 5’-CAAGCAGAAGACGGCATACGAGATACATCGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3’ BC3 5’-CAAGCAGAAGACGGCATACGAGATGCCTAAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3’ BC4 5’-CAAGCAGAAGACGGCATACGAGATTGGTCAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3’ BC5 5’-CAAGCAGAAGACGGCATACGAGATCACTGTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3’ BC6 5’-CAAGCAGAAGACGGCATACGAGATATTGGCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3’ BC7 5'-CAAGCAGAAGACGGCATACGAGATCAGATCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3’ BC8 5’-CAAGCAGAAGACGGCATACGAGATTAGCTTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3’ BC9 5’-CAAGCAGAAGACGGCATACGAGATGATCAGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3’ BC10 5’-CAAGCAGAAGACGGCATACGAGATATCACGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3’ BC11 5'-CAAGCAGAAGACGGCATACGAGATACTTGAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3' BC12 5'-CAAGCAGAAGACGGCATACGAGATGGCTACGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3' BC13 5'-CAAGCAGAAGACGGCATACGAGATCTTGTAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT-3'
- Mckellar, S W, Iosub, I A, Ivanova, I and Granneman, S(2020). Hfq UV cross-linking, ligation and analysis of hybrids (Hfq-CLASH). Bio-protocol Preprint. bio-protocol.org/prep599.
- Iosub, I. A., van Nues, R. W., McKellar, S. W., Nieken, K. J., Marchioretto, M., Sy, B., Tree, J. J., Viero, G. and Granneman, S.(2020). Hfq CLASH uncovers sRNA-target interaction networks linked to nutrient availability adaptation. eLife. DOI: 10.7554/eLife.54655
Category
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.