
- Home
- Protocols

-

Quantification of the diversities of V(D)J recombination events for a given CDR3 peptide
Last updated date: Oct 20, 2020 Views: 1169 Forks: 0
We first conducted multi-color FACS staining of single cell preparations from spleen tissue and peritoneal cavity washout samples. About 1-2 x 104 of phenotypically defined B cells were sorted and RNA was extracted using RNeasy plus Micro Kits (Qiagen) following the manufacturer’s instructions. RNA sample was used for amplicon-rescued multiplex PCR using primers provided by iRepertoire following the procedures that can be found at https://irepertoire.com. About 400bp long PCR products were run on 2% agarose gels and purified using a gel extraction kit (Qiagen). The IgH libraries were pooled and sequenced by Illumina paired-end sequencing (Illumina MiSeq platform). The output of IgH sequence covers CDR2, CDR3 and the beginning of the constant region. The sequence information for all primers used for the library preparation can be found in US Patent Office (US9012148).
Sequence reads were de-multiplexed according to barcode sequences at the 5’ end of reads from the IgH constant region. Reads were then trimmed according to their base qualities with a 2-base sliding window, if either quality value in this window is lower than 20, this sequence stretches from the window to 3’ end were trimmed out from the original read. Trimmed pair-end reads were joined together through overlapping alignment with a modified Needleman-Wunsch algorithm. If paired forward and reverse reads in the overlapping region were not perfectly matched, both forward and reverse reads were thrown out without further consideration. The merged reads were mapped using a Smith-Waterman algorithm to germline V, D, J and C reference sequences downloaded from the IMGT web site(Lefranc, 2003). To define the CDR3 region, the position of CDR3 boundaries of reference sequences from the IMGT database were migrated onto reads through mapping results and the resulting CDR3 regions were extracted and translated into amino acids.
C57BL/6J mouse VH reference sequences were pair-wise aligned with a Smith-Waterman algorithm. Two VH reference sequences are considered related if the aligned region between them is > 200bp matched and < 6 mismatches. Two sequence reads were considered related if the best mapped VH sequences are related and the CDR3 segments have less than 1 mismatch. If two sequences are related and the frequency of the minor one is less than 5% of the dominant one, the minor one is removed from further consideration. In addition, single copy CDR3s are removed from further consideration. To allow multiplexing of multiple samples in a single sequence run, CH primers were linked with barcodes containing 6 different nucleotides. The barcode CH primers were used in a first round RT-PCR. To compensate for potential in chemical synthetic, PCR and/or sequencing error, barcodes were designed with a Hamming distance ≥3. Given that the chemical synthetic error is roughly 5% per position, there is about a 1/8000 chance that one barcode is mistakenly synthesized as another barcode. For a CDR3 with n occurrences in one sample and the same CDR3 (nucleotide sequence) with N occurrences in another sample in the same sequencing run, we calculated the probability that such a CDR3 would occur n or more times if it were due to cross-contamination, using the following formula 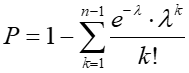
![]() where λ is the expected number of errors given N reads and is computed by λ=N·μ and μ is the cross-contamination rate which is preset as 1/8000. CDR3s that yielded p<0.001 were considered highly unlikely to be due to cross-contamination.
where λ is the expected number of errors given N reads and is computed by λ=N·μ and μ is the cross-contamination rate which is preset as 1/8000. CDR3s that yielded p<0.001 were considered highly unlikely to be due to cross-contamination.
To quantify the diversities of V(D)J recombination events for a given CDR3 peptide, we introduce an entropy value as the index of diversity level. Assuming a distinct CDR3 peptide sequence X in a sample is derived from n number of distinct V(D)J recombinations (nucleotide) with each frequency as P1, P2, … Pn respectively, the entropy for X (Ex) is then calculated as: .
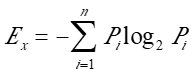
For a sample, after computing entropy values for each distinct peptide CDR3 fragments, the E values for distinct peptide CDR3 fragments are categorized into four ranges: [0, 0.5), [0.5, 1.5), [1.5, 2.5) and [2.5, +∞). The higher the entropy value, the more diverse the V(D)J recombinations for a given CDR3 peptide.
- Han, J and Herzenberg, L A(2020). Quantification of the diversities of V(D)J recombination events for a given CDR3 peptide. Bio-protocol Preprint. bio-protocol.org/prep560.
- Yang, Y., Wang, C., Yang, Q., Kantor, A. B., Chu, H., Ghosn, E. E., Qin, G., Mazmanian, S. K., Han, J. and Herzenberg, L. A.(2015). Distinct mechanisms define murine B cell lineage immunoglobulin heavy chain (IgH) repertoires. eLife. DOI: 10.7554/eLife.09083
Category
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.