
- Home
- Protocols

-

Barcode association of library variants
Last updated date: Jul 20, 2020 Views: 1330 Forks: 0
Barcode labeling of mutant libraries and preparation for paired-end deep sequencing
In this procedure, library variants are tagged with short barcodes via paired-end sequencing to enable tracking of mutations across a large gene using a short sequencing readout. Synthetic DNA constructs containing a randomized 18-bp barcode sequence (N18) are cloned downstream from the gene library of interest via restriction digestion, ligation, and transformation into chemically competent E. coli (Figure 1). The goal is to have each mutant in the library represented by 10-20 unique barcodes. To prepare the barcoded library for paired-end deep sequencing, the distance between the library variant and the barcode is shortened by digestion and recircularization (Figure 2). The resulting DNA is the template for deep sequencing (Figure 3).
Diagram of steps to barcode mutant library
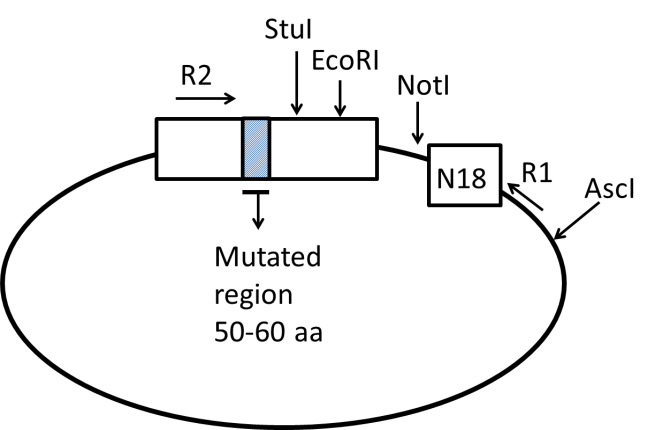
Figure 1. Diagram of barcoded plasmid library. In steps 1-15, N18 barcode cassettes are ligated downstream of the mutated plasmid library. R1 and R2 correspond to Read1 and Read 2 Illumina primers. In our example, the restriction sites NotI and AscI are used for barcode insertion.
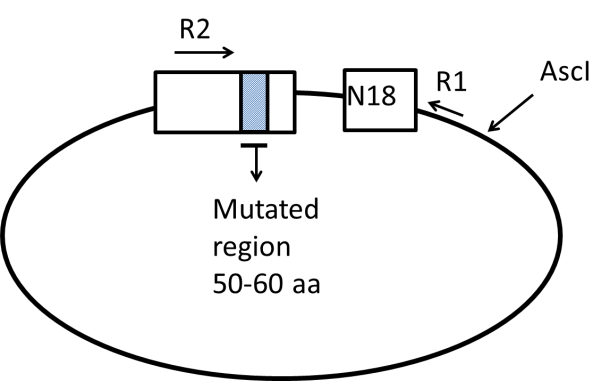
Figure 2. Preparation of the barcoded plasmid library for paired-end sequencing. In steps 16-23, the distance between the mutated region of the library and the N18 barcode is shorted by digestion at the EcoRI and NotI sites and subsequent recircularization of the plasmid.
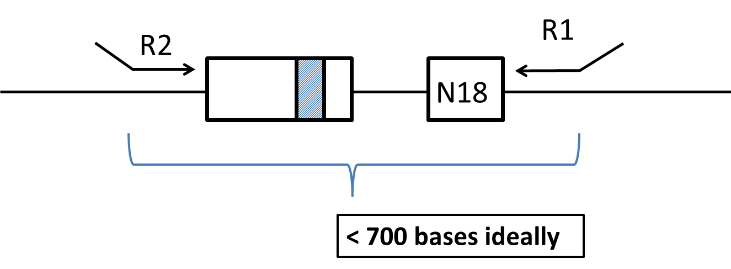
Figure 3: Diagram of linearized plasmid. In steps 24-32 the plasmid is relinearized through digestion by AscI digest to prepare for PCR. The resulting linear DNA is the template for the final PCR reaction that will be used for paired-end deep sequencing.
Reagents
- T4 DNA ligase (New England Biolabs, cat. no. M0202S)
- T4 DNA ligase buffer (New England Biolabs, cat. no. B0202A)
- dNTPs, 10 mM each nucleotide (New England Biolabs, cat. no. N0447)
- Phusion high-fidelity (HF) DNA polymerase (New England Biolabs, cat. no. M0530S, also includes 5x HF buffer)
- Cutsmart NEB buffer (New England Biolabs, cat. no. B7204S)
- NotI HF (New England Biolabs, cat. no. R3189S)
- AscI (New England Biolabs, cat. no. R0558S)
- EcoRI HF (New England Biolabs, cat. no. R3101S)
- T4 DNA polymerase (New England Biolabs, cat. no. M0203S)
- OmniMax competent E coli strain (ThermoFisher Scientific, cat. no. C854003)
- Zymo DNA clean and concentrator-5 kit (Zymo Research, cat. no. D4003)
- ZR Plasmid miniprep classic kit (Zymo Research, cat. no. D4015)
- Calf Intestinal alkaline phosphatase (CIP) (New England Biolabs, cat. no. M0290S)
Table 1. Oligonucleotides
| Oligo name | Oligo sequence (5’ – 3’) | Key features |
| N18 barcode oligo 1 | GGCCGCGAGTCCTGCCNNNNNNNNNNN NNNNNNNAGATCGGAAGAGCGTCGTGG | Anneals with N18 barcode oligo 2 to create N18 barcode cassette. Cassette contains AscI and NotI overhangs specific to plasmid of interest. |
| N18 barcode oligo 2 | CGCGCCACGACGCTCTTCCGATCTNNNNN NNNNNNNNNNNNNGGCAGGACTCGC | Anneals with N18 barcode oligo 1 to create N18 barcode cassette. Cassette contains AscI and NotI overhangs specific to plasmid of interest. |
| Read2 Illumina primer | caagcagaagacggcatacgagatCGGTCTC GGCATTCCTGCTGAACCGCTCTTCCGA TCTTGGCCGCTCTAGAACTAGTAT | Anneals upstream of library in gene of interest. Uppercase sequence will be specific to library. Lowercase represents P7 Illumina sequence. |
| Read1 Illumina primer | aatgatacggcgaccaccgaGATCTACACTC TTTCCCTACACGACGCTCTTCCGATCT | Anneals to N18 barcode cassette. Lowercase region represents P5 Illumina sequence. Underlined region represents sequence that anneals to N18 cassette. |
Procedure for barcoding plasmid libraries
Note: The initial size of the starting library that can be barcoded in one reaction is limited by the efficiency of the ligation of the N18 cassette to the plasmid library and subsequent transformation. We routinely barcode ~4000 variants in one reaction, which requires 40,000-120,000 CFU per ligation reaction.
1. Restriction digestion and dephosphorylation of plasmid library. The restriction sites chosen for barcode insertion should not interfere with transcription termination, but should be located close enough to the gene of interest to facilitate efficient paired-end sequencing. In our example, we inserted the barcode ~200 bp downstream of the gene stop codon. For our specific plasmid, we use NotI and AscI sites to insert the library. Any appropriate compatible sticky end sites can be used. Set up reaction for each library as tabulated below. Incubate for 1 hour at 37°C (or as required for specific enzyme).
| Component | Amount per reaction (µl) | Final |
| Water | 37.5 | |
| Plasmid library (~200 ng/µl) | ~5 | 1 µg |
| Cutsmart NEB buffer (10x) | 5 | 1 x |
| NotI HF (20 U/µl) | 1 | 20 U |
| AscI (10 U/µl) | 1 | 10 U |
| CIP (10 U/µl) | 0.5 | 5 U |
2) Column-purify the digested plasmid using a ZR DNA clean and concentrate kit. Elute in 15 µl elution buffer. Final concentration should be ~ 20-50 ng/µl.
3) Prepare N18 DNA cassettes. Dissolve forward and reverse N18 barcode oligonucleotides in water to a final concentration of 100 µM. Combine 50 µl of forward and 50 µl of reverse oligonucleotides so that the final cassette concentration is 50 µM.
4) Anneal cassettes by boiling followed by slow cooling: float tube containing 100 µl cassettes in beaker of boiling water for 5 minutes, remove water from heat and allow water bath to cool to room temperature (~ 2 hours).
5) Dilute annealed cassettes to 0.1 µM in water.
6) Ligation of plasmid library and N18 cassette. Set up ligation reaction for plasmid and cassette, aiming for an ~ 2:1 ratio of cassette to plasmid concentration and incubate at room temperature for 30 minutes. Include a control without cassette to access the quality of the restriction digest.
| Component | Amount per reaction (µl) | Final |
| Digested plasmid library from step 2 (~10 nM) | 2.5 | ~ 5 nM |
| Annealed cassette from step 5 (100 nM) | 0.5 | 10 nM |
| T4 DNA ligase buffer (10x) | 0.5 | 1x |
| T4 DNA ligase (400 U/µl) | 0.5 | 200 U |
| Water | 1 |
7) Place tubes from step 6 into an ice bath for 5 minutes.
8) Add 100 µl of chemically competent E. coli to each tube. Incubate the tubes on ice for 15 minutes.
9) Place the tubes in a 42°C water bath for 45 seconds, and then place on ice for at least 1 minute. Add 1 ml of room temperature LB broth to each tube and incubate the tubes at 37°C for 1 hour (no longer).
10) To ensure that each mutant is represented by ~10-30 unique barcodes, cultures with different amounts of the transformation reaction are grown overnight and the colony forming units in each culture are assessed by plating a small fraction. Prepare 4 culture tubes with 3 mL LB + selective antibiotic (e.g. LB + ampicillin). Add 25 µl, 50 µl, 100 ul and 200 µl of transformation mix from step 9 into each tube labeled appropriately.
11) To analyze transformation efficiency, plate 30 µl from the tube containing 100 µl of transformed cells from step 10 on a selective LB plate (e.g. LB + ampicillin) and incubate at 37°C overnight.
12) Incubate the four culture tubes from step 10 overnight, rotating at 37°C.
13) The next morning, count the colonies on the plate from step 11 to calculate the transformation efficiency. 50 colonies on the plate would translate to 50,000 independent colony forming units (CFUs) in the original 1 ml of cells from step 9. The target is to choose the tube with a 10-30 fold coverage of the mutants in the plasmid library.
14) Select the tube from step 12 that contains the appropriate number of CFUs and column-purify the culture using a ZR plasmid miniprep kit. Elute in 50 µl elution buffer.
15) To assess the quality of the barcoded library, send for Sanger sequencing.
Preparation of barcoded libraries for paired-end sequencing
16) Restriction digestion to remove DNA between mutant and barcode. In order for efficient paired-end deep sequencing, the region between Read 1 and Read 2 (see Figure 1) should be short, preferably less than 700 base-pairs. If this is not the case, this can be attained by cutting out part of the DNA between the mutated region and the barcode. In our example, there is a NotI cut site just before the barcode, and there are several cut sites such as EcoRI just after the mutated region of the gene. Set up a reaction for each library as tabulated below. Incubate for 1 hour at 37°C (or as required for specific enzyme).
| Component | Amount per reaction (µl) | Final |
| Water | 20 | |
| Barcoded plasmid library (~200 ng/ul) | 5 | 1 µg |
| Cutsmart NEB buffer (10x) | 3 | 1 x |
| EcoRI HF (20 U/µl) | 1 | 20 U |
| NotI (10 U/µl) | 1 | 10 U |
17) Blunt-ending of digested plasmid library. DNA samples are treated with 1 unit of T4 DNA polymerase in the presence of dNTPs. T4 DNA polymerase contains a powerful 3’->5’ exonuclease activity that will remove 5’ overhangs from restriction digestion. Without dNTPs the exonuclease activity would create large single stranded resections. With nucleotides, the polymerase activity counter-balances the exonuclease activity resulting in blunt ends. For enzymes with 3’ overhangs, T4 DNA polymerase will fill in the ends to also create blunt ends. T4 DNA polymerase is very active, and it is important to reign in this activity by using a limited amount of enzyme and performing the reaction at reduced temperature as described in the protocol below.
18) Prepare 1:10 dilution of T4 DNA polymerase. Pipette gently to mix.
| Component | Amount per reaction (µl) | Final | ||
| Water | 8 | |||
| Cutsmart NEB buffer (10x) | 1 | 1 x | ||
| T4 DNA polymerase (3 U/µl) | 1 | 0.3 U/µl | ||
19) Prepare 12°C water bath using approximately 100 mL ice in 900 mL room temperature water.
20) Prepare restriction-digested sample from step 16 for blunt ending as below.
| Component | Amount per reaction (µl) | Final | |
| Digested sample from step 16 | 30 | ||
| Cutsmart NEB buffer (10x) | 3.4 | 1 x | |
| dNTP (10mM) | 0.8 | 0.3 mM | |
21) Float your sample from step 20 in the 12°C water bath for 1 minute to temperature equilibrate.
22) Add 3 µL of 1:10 diluted T4 DNA polymerase mix from step 18 and pipette gently to mix. Incubate at 15 minutes in the 12°C water bath. Proceed to column purification immediately (it is important to remove the T4 DNA polymerase at this point).
23) Column-purify the digested and blunt-ended plasmid using a ZR DNA clean and concentrator kit. Elute in 15 µl elution buffer.
24) Ligation to re-circularize plasmid. Ligation is performed at low concentration (~ 3ng/ul) so that circularization outcompetes bi-molecular ligations. Set up ligation reaction as tabulated below. Leave at room temperature overnight.
| Component | Amount per reaction (µl) | Final |
| Digested plasmid library from step 23 (~60 ng/µl) | 5 | ~ 300 ng |
| T4 DNA ligase buffer (10x) | 10 | 1x |
| T4 DNA ligase (400 U/µl) | 2.5 | 1000 U |
| Water | 82.5 |
25) Column-purify the re-circularized plasmid from step 24 using a ZR DNA clean and concentrator kit. Elute in 15 µl elution buffer. The final product should be ~20 ng/µL.
26) Linearization of plasmid. To maximize the efficiency of PCR the template should be linear. The circularized plasmid should be re-linearized again while keeping the barcode and mutated region adjacent to each other. The plasmid should be cut before the mutated region or after the barcode, not in between (see Figure 3). Set up the restriction digest as tabulated below. The exact amount of DNA used for this reaction is not critical. Digest at 37°C for 1 hour. No need to column purify before proceeding to PCR step.
| Component | Amount per reaction (µl) | Final |
| Water | 3 | |
| Barcoded plasmid library (~20 ng/µl) | 5 | 100 ng |
| Cutsmart NEB buffer (10x) | 1 | 1 x |
| AscI (10 U/µl) | 1 | 10 U |
27) Test PCR to check quality of library to prepare for paired-end sequencing run. Set up PCR reaction with the following:
| Component | Amount per reaction (µl) | Final |
| Template (~10 ng/µl) | 1 | ~10 ng |
| Read 1 primer (5 µM) | 2 | 0.5 µM |
| Read 2 primer (5 µM) | 2 | 0.5 µM |
| dNTP (10 mM) | 0.5 | 0.25 µM |
| 5x HF buffer | 4 | 1x |
| Phusion | 0.5 | |
| Water | 10 |
28) Run the samples in a thermocycler with the following conditions:
| STEP | TEMP | TIME |
| Initial denaturation | 98°C | 30 seconds |
| 10-15 cycles | 98°C 55°C 72°C | 10 seconds 30 seconds 90 seconds |
| Final extension | 72°C | 5 minutes |
| Hold | 4°C |
29) Analyze 10 ul of your PCR product on a 1.2% agarose gel. Look for a clear but faint band at the correct size. If band is bright, reduce number of cycles as to not saturate the PCR reaction. If band is correct, proceed to final PCR step.
30) Amplification of library for paired-end sequencing run. Set up two PCR reactions with the following components:
| Component | Amount per reaction (µl) | Final |
| Template (~10 ng/µl) | 5 | ~50 ng |
| Read 1 primer (5 µM) | 5 | 0.5 µM |
| Read 2 primer (5µM) | 5 | 0.5 µM |
| dNTP (10 mM) | 1 | 0.2 µM |
| 5x HF buffer | 10 | 1x |
| Phusion | 1 | |
| Water | 23 |
31) Run the samples in a thermocycler with the same conditions and cycles as determined for step 28.
32) Combine both PCR reactions and column purify using the ZR DNA clean and concentration kit. Elute in 15 µl. Perform a second column purification to ensure removal of primers that would interfere with sequencing, elute in 15 µl, check concentration, and store at -20C. Sample is now ready for Illumina paired-end sequencing run.
- Flynn, J and Bolon, D(2020). Barcode association of library variants. Bio-protocol Preprint. bio-protocol.org/prep403.
- Flynn, J. M., Rossouw, A., Cote-Hammarlof, P., Fragata, I., Mavor, D., Hollins, C., Bank, C. and Bolon, D. N.(2020). Comprehensive fitness maps of Hsp90 show widespread environmental dependence. eLife. DOI: 10.7554/eLife.53810
Category
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.