
- Home
- Protocols

-

Differentiation of rTTA/Ngn2-positive PSCs to iNeurons
Last updated date: May 22, 2023 DOI: 10.21769/p2308 Views: 1079 Forks: 0
iPSCs-derived Ngn2-induced neurons
Maria Schörnig1, Valentina Rava2 and Elena Taverna2
1 Institute for Biochemistry, Universitätsklinikum Jena, Jena, Germany
2 Human Technopole, Milan, Italy
1. Glia Cell Isolation and Culture
To obtain embryonic (E18) rat brains:
- Sacrifice an E18 pregnant rat.
- Harvest the embryos from the uterus, isolate the brains.
Use the cortices from two embryonic brains to fill a T75 flask. The cortices can be combined in one tube for the dissociation.
Prepare flasks
1. Dilute Poly-D-Lysine (PDL) in sterile water (final concentration 10 µg/mL). Cover the entire surface of the flask by adding 5 mL of PDL. Place the flask for 1-2 h in a humidified 37 °C incubator. The flask can be also stored at 4 °C for 1-2 days before use.
2. Remove PDL from the flask. Rinse the flask 3x with 5 mL of sterile PBS to remove excess PDL. Aspirate PBS and use immediately, to avoid the PDL-coated surface to dry, by adding 5 mL of astrocyte culture medium.
Dissection of the brain tissue cortices
3. Prepare 50 mL of dissection medium: Lebovitz's L-15 medium supplemented with 2% (v/v) B-27. Keep on ice.
4. Sacrifice the rat by deep sedation followed immediately by euthanasia by cervical dislocation.
5. Spray 70% EtOH over the rat’s abdomen, wipe carefully. Cut the skin and open the abdomen via Caesarean section. Expose and remove the uterus using scissors.
6. Remove the embryos from the amniotic sacs, transfer to a sterile Petri dish and immediately decapitate using scissors.
7. Transfer the heads to a sterile 50 mL falcon tube filled with cold dissection medium.
8. Extract the brains from the embryos under a stereo microscope using forceps (n. 5). Remove the skin and skull using forceps (n. 55). Collect the brains and transfer to a 35 mm Petri-dish containing cold dissection medium.
9. Remove completely the meninges using forceps (n. 55). Using spring scissors separate the two hemispheres. When the two hemispheres are separated, check if all meninges are removed from the medial part of each of them. If not, proceed with a second round of meninges removal, using forceps (n. 55).
10. Using a scalpel or spring scissors, remove the olfactory bulb, the midbrain, the striatum, and the hippocampus.
11. Cut the cortices into smaller pieces using a scalpel.
12. Collect the cortices in a 15 mL centrifuge tube filled with 5 mL of cold dissection medium. Place on ice.
Dissociation of the cortices
13. Prepare 2 mL of Ca2+/Mg2+-free Hank's Balanced Salt Solution (HBSS) with 0.25% trypsin (dissociation medium). Prepare 50 mL of high-glucose Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 15% (v/v) Fetal Bovine Serum (FBS) and 1% (v/v) penicillin/streptomycin (culture medium), and filter sterilize.
14. Wash the tissue with Ca2+/Mg2+-free HBSS (without trypsin), after the tissue has settled, remove the HBSS.
15. Add 2 mL of dissociation medium. Move the tube gently by flicking, to allow the enzyme mix to homogeneously cover the tissue.
16. Incubate in a water bath at 37 °C for 10 min. Flick the tube very gently few times during incubation.
17. Using a 1,000 µL pipette tip, triturate the tissue by aspirating (and ejecting) the mixture 2 times. Add as quickly as possible 1 mL of culture medium to inactivate the trypsin.
18. Proceed with trituration by aspirating (and ejecting) the mixture 10 times. Check if the tissue is dissociated. If not, repeat aspiration and ejection for max additional 3 times.
19. Add 7 mL of culture medium to inactivate the trypsin and gently mix the tube by flicking it.
20. Pass the cell suspension through a 70 µm cell strainer previously placed on a 50 mL centrifuge tube. Rinse the 15 mL tube with culture medium and filter the medium through the cell strainer to collect it in the tube with the cell suspension. Rinse the cell strainer a few times with culture medium.
21. Centrifuge at 200 x g for 10 min to pellet the cells. Aspirate the medium without touching the pellet. Resuspend the pellet in 1 mL of culture medium. Add 10 mL of prewarmed culture medium. Mix the cell suspension.
22. Rinse the PDL-coated flask with culture medium. Remove the medium. Transfer the cell suspension into the flask.
23. Start astrocytes culture by placing the flask into a humidified 37 °C incubator, 5% CO2.
Expansion and maintenance of the astrocytes
24. Replace the entire medium after 2 days. After that, replace the medium every 3 days. The medium should always be prewarmed before adding it to the cells.
25. When the astrocytes are approximately 90% confluent, remove contaminating glial cells (mainly microglia and oligodendrocytes) by using an orbital shaker:
1) Take the flask out from the incubator. Close the cap. To remove microglia, shake the flask on an orbital shaker set at 180 rpm for 1 h. Remove the medium and rinse the flask twice with warm culture medium. Add 12 mL of fresh culture medium.
2) To remove the oligodendrocytes, shake the flask at 250 rpm, and incubate at 37 °C for a minimum of 7 h or, preferably, O/N.
3) Remove the medium and rinse the flask twice with warm culture medium. Add 12 mL of fresh culture medium.
4) When 100% confluent, split the astrocytes using standard procedures with 0.05% trypsin-ethylenediaminetetraacetic acid (EDTA). Astrocytes should be diluted at 1:3 to 1:2.
Cryopreservation of astrocytes
26. Astrocytes can be stored by freezing them in freezing medium.
27. Thaw astrocytes using standard procedures.
Although fresh astrocytes might work best, we did obtain indistinguishable results by using frozen astrocytes prepared in house or purchased by a vendor.
2. Generation of rtTA/Ngn2-positive pluripotent stem cell lines
We used human, chimpanzee, and bonobo iPS cell lines and one additional human ES cell line.
- The human iPSCs line HmRNA (generated in this study), coming from human dermal fibroblasts, was reprogrammed using the StemMACS mRNA transfection kit.
- The validation of the cell line for pluripotency markers was performed by immunohistochemical staining using the Human Pluripotent Stem Cell 3-Colour Immunohistochemistry Kit.
- Cells were differentiated into the three different germ layers using the Human Pluripotent Stem Cell Functional Identification kit and StemMACS Trilineage Differentiation Kit.
- Karyotyping was done using Giemsa banding. Karyotypes were found to be normal. The human ES cell line H9 was purchased from WiCell.
- The chimpanzee iPSC lines SandraA and JoC as well as the bonobo iPSCs line BmRNA were generated in a previous study (Kanton et al. 2019).
- The rtTA/Ngn2-positive iPSCs/ESCs were generated using lentiviral vectors to stably integrate the transgenes into the genome of the pluripotent stem cells, as previously described by Frega et al. (Frega et al. 2017).
2.1 Viral transduction for creating rtTA/Ngn2-positive iPS-cell lines
Day 0: Plate iPSCs as single cells
Preparations:
- Prepare a geltrex-coated 12-well plate: add 500 μL of geltrex per well. For one cell line 4 coated wells of a 12-well plate are needed.
- Prewarm the required volume of mTeSR™1 medium supplemented with ROCK inhibitor (final concentration 10 µM) at RT.
- Prewarm the required volume of DMEM/F12 to 37 °C. DMEM/F12 is used for washing the cells after accutase treatment. For plating one iPSC line, 11 mL of DMEM/F12 are sufficient.
- Prewarm the required volume of accutase to 37 °C.
Protocol:
- Add 1 mL of accutase to the iPS cells (in case of 6-well plate). Incubate from 3 to 5 min at 37 °C and check under the microscope whether the cells are detaching from one another (“rounding up”).
- Add 2 mL of DMEM/F12, gently suspend the cells and transfer to a 15 mL tube.
- Add 8 mL of DMEM/F12 to the cell suspension.
- Centrifuge at 1500 rpm for 5 min at RT.
- Aspirate the supernatant and resuspend the cells in 2 mL of previously prepared mTeSR™1 medium. Resuspend the cell pellet as single cells by putting the tip of the pipette against the side of the 15 mL tube and resuspending the cells gently but vigorously.
- Count the cells using a counting chamber.
- Aspirate the geltrex and plate the dissociated cells in the prepared 12-well plate at a density of 30,000 cells per well.
- Incubate at 37 °C, 5% CO2.
Day 1: Transduce the iPS cells with lentiviral constructs
The pLVX-EF1α-(Tet-On-Advanced)-IRES-G418(R) (rtTA) and pLVX-(TRE-thight)- (MOUSE)Ngn2-PGK-Puromycin(R) (Ngn2) vectors were provided by Nael Nadif Kasri´s lab (Frega et al. 2017). The virus particles were produced by the viral core facility of the Charité Medical Hospital in Berlin, Germany. The virus particles were harvested in DMEM medium with approximately 106 particles per mL.
Preparations:
- Prewarm the required volume of mTeSR™1 medium to 37 °C. Add polybrene to a final concentration of 8 μg/mL (dilute stock 1:1000) and add ROCK inhibitor (final concentration 10 µM).
- Thaw the aliquots of the lentivirus on ice.
- After 6 hours incubation: prewarm the required volumes of mTeSR™1 medium.
Protocol:
1. Aspirate the spent medium.
2. Add to each well 1 mL of ROCK-inhibitor-supplemented mTeSR™1 medium.
3. Per iPS cell line for which a rtTA/Ngn2-positive line needs to be generated, perform the transduction with different amounts of lentivirus. That is, perform three transductions in technical duplicates per iPS cell line:
- Transduction 1: add 100 μL of each lentiviral vector (rtTA + Ngn2).
- Transduction 2: add 200 μL of each lentiviral vector.
- Transduction 3: add 300 μL of each lentiviral vector.
The non-transduced wells will serve as controls for the selection.
4. Incubate at 37 °C, 5% CO2 for 6 hours.
5. After 6 hours: aspirate the spent medium. Add 1 mL of mTeSR™1 medium to each well.
6. Incubate at 37 °C, 5% CO2 O/N.
Day 2: Refresh the mTeSR™1 medium.
Preparations:
1. Prewarm the required volume of mTeSR™1 medium to 37 °C.
Protocol:
- Aspirate the spent medium.
- Add 1 mL of the prepared mTeSR™1 medium to each well.
- Incubate at 37 °C, 5% CO2 O/N.
Day 3-7: Selection with G418 and puromycin
As an alternative to puromycin, a vector with a blasticidin resistance can be used coupled with the selection against the given antibiotic.
1. Prewarm the required volume of mTeSR™1 medium to 37 °C (in case some wells need to be split, prepare extra medium). Add the antibiotics required for the selection. The amount of the antibiotics differs according to the selection period:
Table 1: Concentration of G418 antibiotic according to the day of selection
Day | Final concentration of G418 | Dilution of G418 stock solution (50 mg/mL) |
Day 3 | 100 µg/mL | 1:500 |
Day 4-7 | 250 µg/mL | 1:200 |
Table 2: Concentration of puromycin antibiotic according to the day of selection
Day | Final concentration of puromycin | Dilution of puromycin stock solution (1 mg/mL) |
Day 3 | 1 µg/mL | 1:1000 |
Day 4 | 2 µg/mL | 1:500 |
Day 5 | 2 µg/mL | 1:500 |
Day 6 | 1 µg/mL | 1:1000 |
Day 7 | 0.5 µg/mL | 1:2000 |
2. In case some wells need to be split, prewarm the required volumes of DPBS and EDTA to RT. In addition, coat 6-well plates with matrigel (as described in the section “Splitting iPS cells”).
Protocol:
- Aspirate the spent medium.
- Add 1 mL of the prepared mTeSR™1 medium (i.e., supplemented with the antibiotics for selection) to the wells that don’t need to be split.
- Split 70-80% confluent cells, according to protocol section “Splitting iPS cells”. Use instead of the normal mTeSR™1 medium, the mTeSR™1 medium supplemented with antibiotics for selection and ROCK inhibitor (final concentration 10 µM). Transfer all the split cells of one well (in case of a 12-well plate) to one well of a 6-well plate.
- Incubate at 37 °C, 5% CO2 O/N.
Day 8: Stop the selection and start regular culturing.
After selecting the rtTA-positive and Ngn2-positive iPS cells, the cells can be cultured as described, in mTeSR™1 medium supplemented with G418 (final concentration 50 μg/mL) and puromycin (final concentration 0.5 μg/mL) (dilute stocks 1:1000 and 1:2000, respectively). The cells should be expanded for freezing (backup).
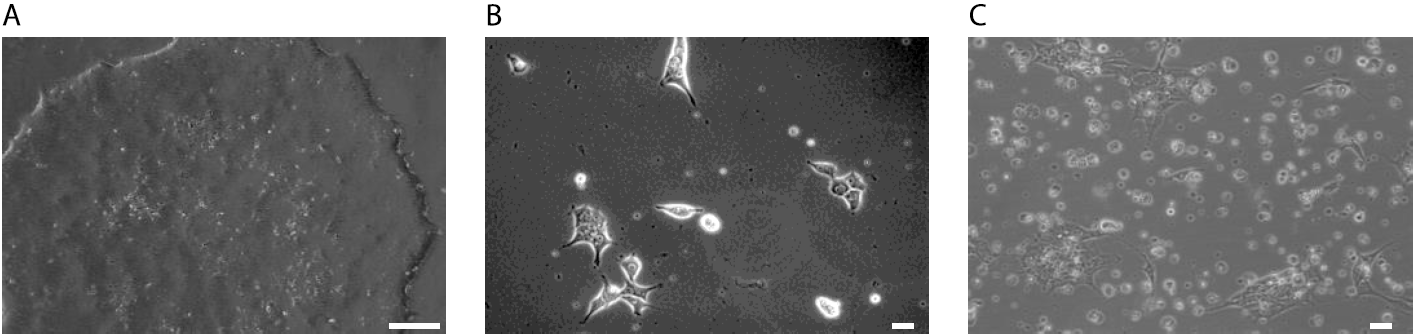
Figure 1: Selection of rtTA-Ngn2 transduced iPS cells. A: iPSCs grow as densely packed colonies before selection. Scale bar: 2.5 cm B: iPSCs 6 h after transduction with lentivirus. iPSCs were seeded as single cells. Morphological changes are possible due to virus transduction and incubation with reagents needed for the transduction, like polybrene. Scale bar: 10 µm. C: iPSCs during the selection with antibiotics. It is common to observe a lot of cell death. Selected iPSCs will grow again as densely packed colonies and need to be split when they reach 60-80% confluency. Scale bar: 10 µm.
3. Culture of pluripotent stem cell lines
- Perform the culturing of human, chimpanzee, and bonobo rtTA/Ngn2-positive pluripotent stem cells in 6‑well cell culture plates coated with Matrigel (1:100 dilution in Knockout DMEM/F12). Regularly test the cultures for mycoplasma.
- Maintain rtTA/Ngn2-iPSCs in mTeSR™1 medium with supplement and antibiotics (final concentration: Pen/Strep 50U/mL, G418 50µg/mL, and Puromycin 0.5 µg/mL) at 37 ˚C, 5 % CO2.
- Refresh (change) the medium every day.
- To maintain the culture, either pick and seed pluripotent stem cell colonies with a round and densely packed morphology into wells containing fresh medium or split once they are approximately 80 % confluent.
- To split, wash the cells with DPBS before incubation with 1ml of EDTA (final concentration 0.5mM, diluted in PBS) for 5 min at 37 °C. Aspirate EDTA and detach the cells from the culture dish with 1ml medium. After splitting, transfer the cell suspension into new wells containing fresh mTeSR™1 with supplement. Add ROCK Inhibitor (10µM final concentration) to the culture medium and keep it for 24 h.
4. Cryopreservation of pluripotent stem cells
- To collect cells for freezing wash them with DPBS twice before incubation with 1 mL TrypLE™ Express for 5 min at 37 °C.
- Wash the cells from the culture dish with 3 mL of DMEM/F12 and centrifuge at 200 x g for 5 min.
- Resuspend the pellet in 500 µL of mFreSR™, and transfer it into a cryogenic vial, gradually freeze to -80 °C in an isopropanol filled freezing box.
5. Thaw iPSCs
- To thaw iPSCs, quickly put the cryogenic vial in the water bath at 37 °C for a maximum of 2 min.
- Transfer the cells, with a wet pipet tip, into a falcon already containing 5ml of DMEM/F12.
- Centrifuge the cell suspension for 5 min at 200 x g.
- After centrifugation, discard the supernatant, and resuspend the cell pellet with 1 mL of iPSCs culture medium with ROCK Inhibitor (final concentration 10µM).
- Plate 1/3 and 2/3 of cells into two new wells (previously coated with Matrigel) in a final volume of 2 ml iPSCs culture medium supplemented with ROCK Inhibitor (final concentration 10 µM).
6. Differentiation of rTTA/Ngn2-positive pluripotent stem cells into iNeurons
Human, chimpanzee, and bonobo pluripotent stem cells were differentiated into induced Neurons (iNs) according to Frega et al., 2017 protocol (Frega et al. 2017). iPSCs or ESCs were plated as single cells and the neuronal differentiation was initiated by doxycycline - induction of Ngn2. In brief:
- Plate iPSCs or ESCs as single cells and initiate the neuronal differentiation by doxycycline-induction of Ngn2.
- Coat pre-coated acid-treated coverslips with 50 µg/mL of PLO in borate buffer for at least 3 h at 37°C. Wash the coverslips three times with sterile MQ H2O and coat them with 10 µg/mL mouse laminin in DMEM/F12; leave O/N at 4°C.
- Plate the cells as single cells in mTeSR™1 medium supplemented with 1% P/S, 4 µg/mL doxycycline and ROCK inhibitor (final concentration 10 µM). Filter the culture medium through a 0.22 µm filter.
- On day one of differentiation, change the culture medium to DMEM/F12 supplemented with 1% P/S, N2-supplement (final concentration 1% v/v), non-essential amino acids (NEAA) (final concentration 1% v/v), NT-3 (final concentration 10 ng/mL), BDNF (final concentration 10 ng/mL), doxycycline (final concentration 4 µg/mL) and mouse laminin (final concentration 0.2 µg/mL).
- After two days of culturing add primary cortical rat astrocytes to the system.
- One day after astrocyte plating, change the culture medium to Neurobasal medium supplemented with 1% P/S, B-27 supplement (final concentration 2% v/v), glutamax (final concentration 1% v/v), doxycycline (final concentration 4 µg/mL), NT-3 (final concentration 10 ng/mL) and BDNF (final concentration 10 ng/mL).
- Stop proliferation of undifferentiated pluripotent stem cells and proliferating astrocytes by supplementing the growth medium with 2 µM cytosine arabinoside (Ara-C) on day three of cell culture.
- From day 10 onwards, additionally supplement the Neurobasal growth medium with 2.5% fetal bovine serum (FBS), to promote the rat astrocytes.
- Keep iNs in culture for two to eight weeks. Change 50% of the medium every two to three days.
For a detailed protocol, please follow the description below.
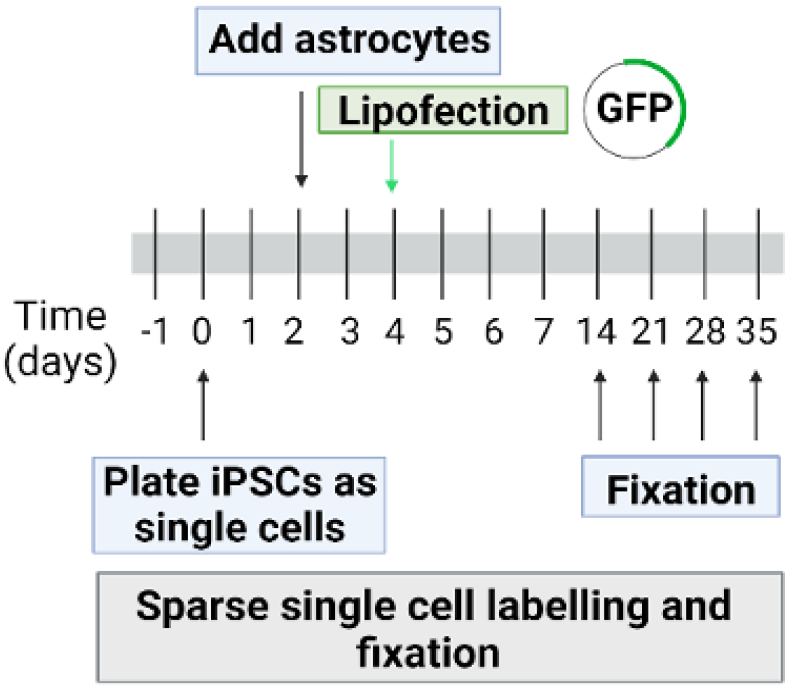
Figure 2: Schematic description of iNeurons differentiation protocol (based on (Schörnig et al. 2021)).
6.1 Day -1. Coat with Poly-L-Ornithine and laminin
Sterilize 12 mm glass coverslips with 3 washings in 70% ethanol and dry them under UV light for 30 min. Two distinct protocols can be used to coat 12 mm coverslips used for the induction of iPSCs differentiation:
- Coat the treated coverslips with 500 µL of Poly-L-Ornithine (Sigma-Aldrich) (PLO) (final concentration 50 µg/mL, diluted in Borat buffer (4.76 g Boric acid, 2.54 g Borax in 1 L of distilled water)). While adding the PLO, keep the coverslip down with a p1000 tip to avoid fluctuation. Incubate the coverslips with PLO overnight at 37 °C, 5% CO2. The following day discard PLO and wash the coverslips with ultrapure distilled water for three times. To coat the coverslips with laminin, add 500 µL of Laminin (Sigma- Aldrich) (final concentration 10 µg/mL diluted in DMEM/F12) to each coverslip and incubate at 37°C for 2 h.
- Coat the sterilized coverslips with 500 µL of PLO (final concentration 50 µg/mL). Incubate the coverslips with PLO for at least 3 h at 37 °C, 5% CO2. Afterwards discard PLO and wash the coverslips with ultrapure distilled water for three times. Add 500 µL of Laminin (final concentration 10 µg/mL) to each coverslip and incubate overnight at 4 °C.
6.2 Day 0. Plate iPSCs
- To detach iPSCs from the 6 well plate, discard the culture medium and wash once with DPBS -/-.
- Incubate cells with 1 mL of accutase (Sigma-Aldrich) for 5 min at 37 °C.
- Wash iPSCs with 2 mL of DMEM/F12, gently pipetting with a p1000 to transfer the cells to a falcon.
- Centrifuge iPSCs at 200 x g for 5 min at RT.
- Discard the supernatant and resuspend the pellet by pipetting gently up and down, to obtain iPSCs as single cells, with 1 mL of new medium containing mTeSR™1 supplemented with P/S (final concentration 50 U/mL), Doxycycline (Sigma-Aldrich) (final concentration 4 µg/mL, reconstitution in ultrapure distilled water), and ROCK Inhibitor (final concentration 10 µM).
- Count the cells and dilute them in the culture medium to reach a concentration of 100,000 iPSCs/mL. Plate cells on previously coated coverslips, and culture at 37 °C, 5% CO2.
6.3 Day 1. Media change
- Prepare the differentiation medium containing DMEM/F12 supplemented with N2 (Thermofisher) (final concentration 1% v/v), P/S (final concentration 50 U/mL), MEM Non-Essential Amino Acids Solution (Thermofisher) (final concentration 1% v/v), Doxycycline (final concentration 4 µg/mL), Rock Inhibitor (final concentration 10 µM), Recombinant Human NT3 (NT3) (Peprotech) (final concentration 10 ng/mL), and Recombinant Human/Murine/Rat BDNF (BDNF) (Peprotech) (final concentration 10 ng/mL).
- Filter the medium through a 0.22 µm filter, then add mouse laminin (final concentration 0.2 µg/mL).
- Wash cells once with DPBS -/- and add 500 µL of fresh prewarmed (at 37 °C) differentiation medium.
6.4 Day 2. Add rat astrocytes
The second day of differentiation add rat astrocytes to iNeurons culture.
Maintain rat primary cortical astrocytes (Thermofisher) culture in T75 flasks, coated with Poly- D-Lysine (Sigma-Aldrich) (final concentration 10 µg/mL), in growth medium composed by DMEM (Thermofisher) supplemented with 15% Fetal Bovine Serum (FBS) (Sigma-Aldrich), and P/S (final concentration 50 U/mL). To co-culture astrocytes with iNeurons:
- Wash the astrocytes with DPBS -/- and incubate them with 5 mL of TrypLE™ Express for 5 min at 37°C.
- Wash astrocytes with the spent medium to stop the enzymatic reaction.
- Centrifuge the cells at 250 x g for 5 min.
- Discard the supernatant and resuspend the pellet with 1 mL of new astrocytes medium.
- Count the cells, and plate them as single cells on iNeurons with a final concentration of 40,000 astrocytes/mL. Continue the co-culture at 37 °C, 5% CO2.
6.5 Day 3. Media change
- Prepare the differentiation medium containing Neurobasal medium (Thermofisher) supplemented with B27 Supplement (B27) (Thermofisher) (final concentration 2% v/v), P/S (final concentration 50 U/mL), GlutaMAX™ Supplement (GlutaMAX) (Thermofisher) (final concentration 1% v/v), Doxycycline (final concentration 4 µg/mL), Cytosine β-D-arabinofuranoside (AraC) (Sigma-Aldrich) (final concentration 2 µM), NT3 (final concentration 10 ng/mL), and BDNF (final concentration 10 ng/mL). Filter the medium through a 0.22 µm filter.
- Wash the cells once with DPBS -/- to remove dead astrocytes and add 500 µL of fresh prewarmed (at 37 °C) differentiation medium.
6.6 Day 6 and 8. 50% media change
From the sixth day in vitro change only 50% of the medium; therefore, discard 250 µL of old medium, and add 250 µL of prewarmed fresh differentiation medium to the iNeuron culture. The differentiation medium contains Neurobasal supplemented with B27 (final concentration 2% v/v), P/S (final concentration 50 U/mL), GlutaMAX (final concentration 1% v/v), Doxycycline (final concentration 4 µg/mL), NT3 (final concentration 10 ng/mL), and BDNF (final concentration 10 ng/mL). Filter the medium through a 0.22 µm filter.
6.7 From day 10 on. 50% media change
From the tenth day in vitro on, change only 50% of the medium. The differentiation medium contains Neurobasal supplemented with 2.5% FBS, B27 (final concentration 2% v/v), P/S (final concentration 50 U/mL), GlutaMAX (final concentration 1% v/v), Doxycycline (final concentration 4 µg/mL), NT3 (final concentration 10 ng/mL), and BDNF (final concentration 10 ng/mL). Filter the medium through a 0.22 µm filter.
7. Lipofection of iNeurons
To visualize iNs soma and neurites at the single cell level, we developed a sparse labelling method for iNeurons. Plate cells on 12 mm coverslips and lipofect (transfect) them with a plasmid encoding cytoplasmic GFP (pCAGG1-GFP) at day 4 (d4) in vitro using the Lipofectamine™ 3000 Transfection Reagent (Thermofisher). The protocol used is as follows: Prepare two different mixes: lipofectamine mix, and DNA mix
Table 3: Lipofection reaction mix
Mix |
Volume |
|
Mix |
Volume |
OptiMEM (Thermofisher) | 25 µL | Optimem | 25 µL | |
Lipofectamine 3000 reagent | 0.75 µL | pCAGG1-GFP | 0.1 µg | |
| P3000 reagent | 1 µL | ||
Add the DNA mix to the lipofectamine mix and incubate for 15 min at RT. Add 50 µL of the mix to iNeurons and put the cells back in the incubator at 37 °C, 5% CO2 for 48 h following which the differentiation media needs to be completely changed.
iNs were then kept in culture until fixation at different time points (d14, d21, d28, and d35) to follow neuronal maturation and synaptogenesis over time at the single cell level. In parallel, we also kept in culture non-lipofected iNeurons to check for neuronal markers expression.
![]()
![]()
![]()
![]()
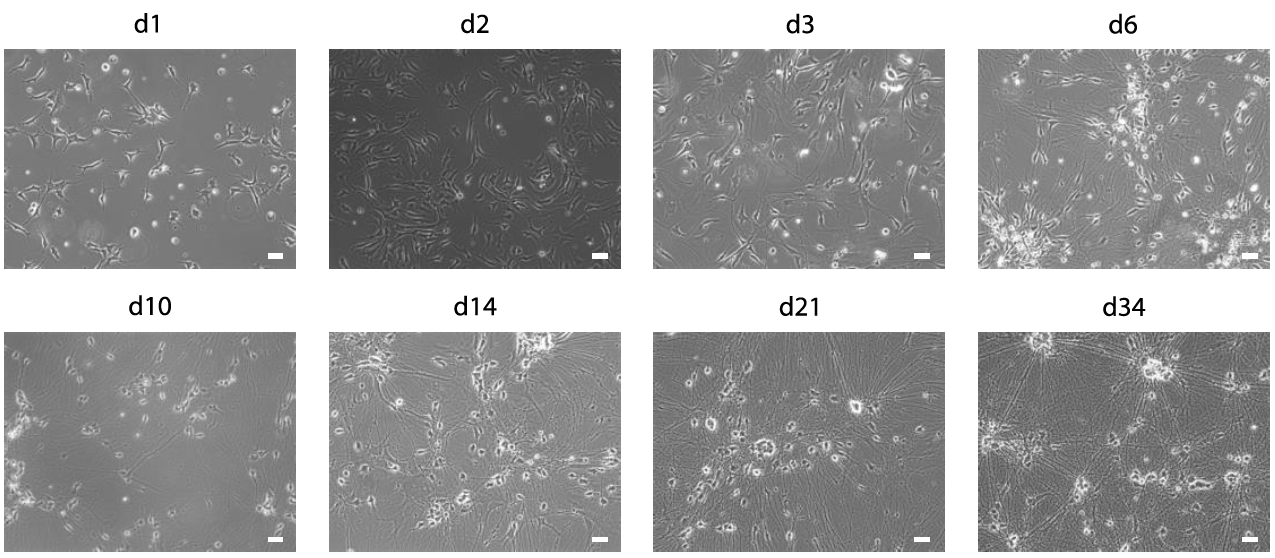
Figure 3: Differentiation of iNeurons. Phase contrast images of human iNeurons (cell line hiPS-409-B2_Ngn2) during differentiation at d1, d2, d3 (before astrocyte plating), d6 (3 days after astrocyte plating), d10, d14, d21 and d34. Scale bars: 10 µm.
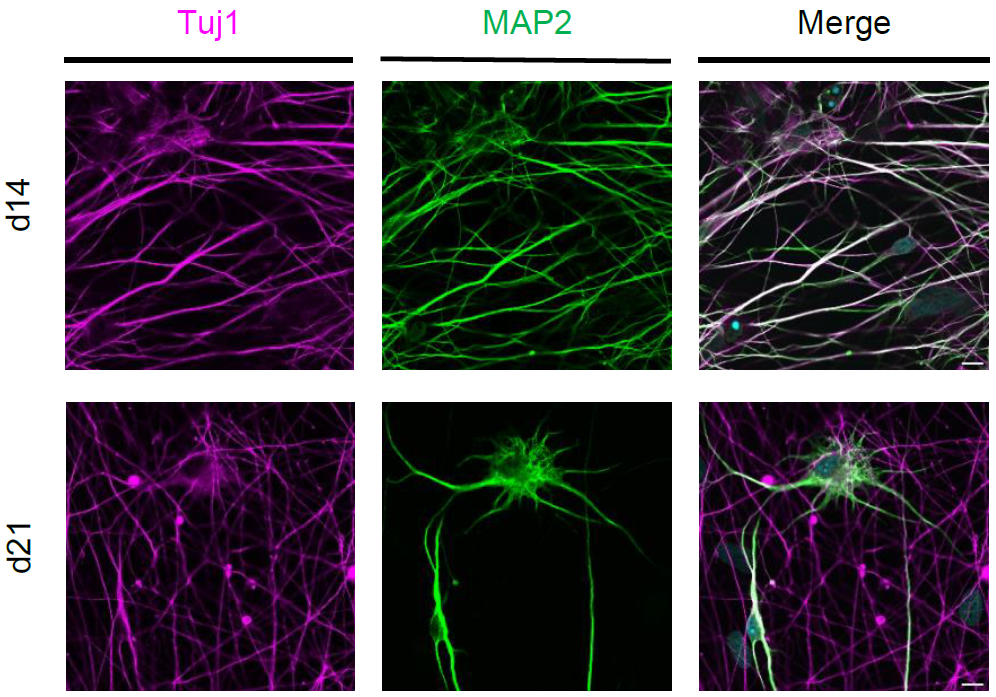
Figure 4: Characterization of iNeurons. Confocal images of human iNeurons (cell line hiPS-409-B2_Ngn2) after fixation at d14 and d21. Immunofluorescence with neuronal markers: Tuji1 (magenta) and MAP2 (green). Scale bars: 10µm.
Supplementary Figures:
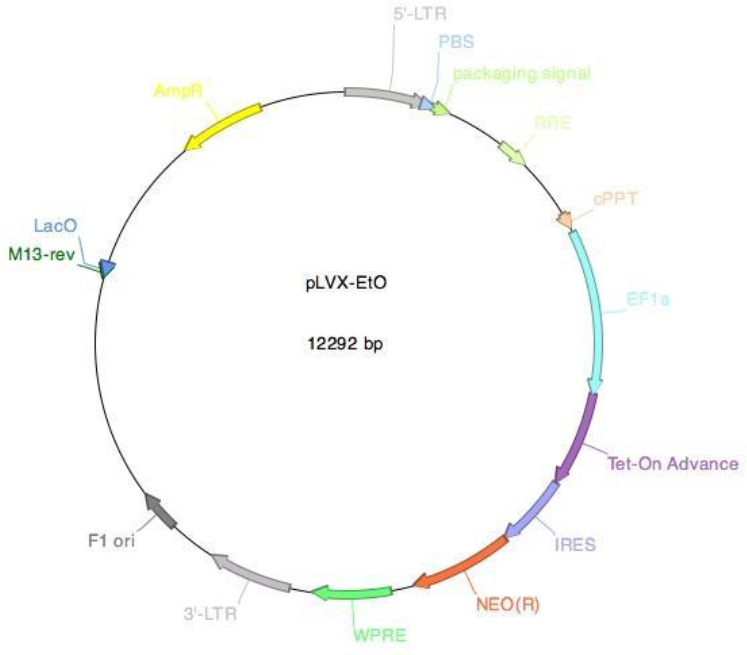
Supplementary Figure 1: Vector card for the pLVX-EtO vector. The pLVX-EtO vector (pLVX-EF1α-(Tet-On- Advanced)-IRES-G418(R)) encodes the Tet-On advanced transactivator (rtTA) for the doxycycline inducible system.
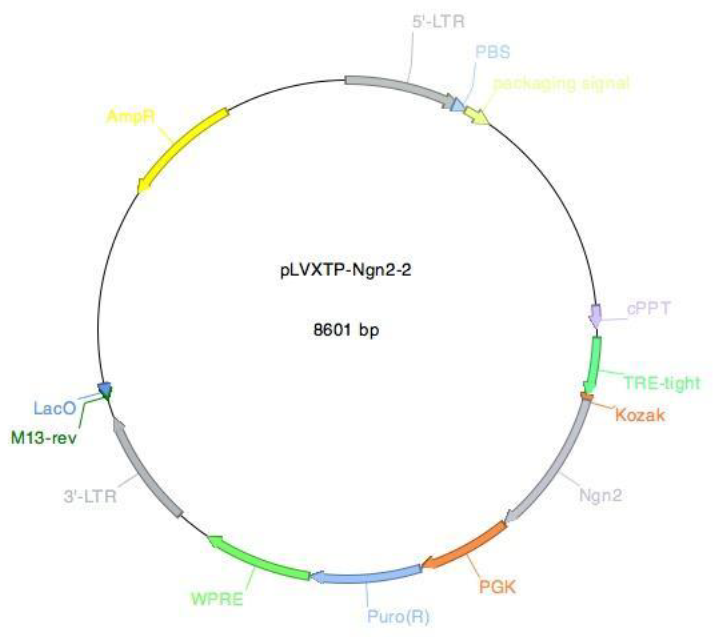
Supplementary Figure 2: Vector card for the pLVXTP-Ngn2 vector. The pLVXTP-Ngn2 vector (pLVX-(TRE- thight)-(MOUSE)Ngn2-PGK-Puromycin(R)) encodes the murine Ngn2 gene. The expression of mouse Ngn2 is controlled by the rtTA-protein bound to doxycycline in this system.
References:
Frega, M., S. H. van Gestel, K. Linda, J. van der Raadt, J. Keller, J. R. Van Rhijn, D. Schubert, C. A. Albers, and N. Nadif Kasri. 2017. 'Rapid Neuronal Differentiation of Induced Pluripotent Stem Cells for Measuring Network Activity on Micro-electrode Arrays', J Vis Exp, 119: 1-10 e54900
Kanton, S., M. J. Boyle, Z. He, M. Santel, A. Weigert, F. Sanchis-Calleja, P. Guijarro, L. Sidow, J. S. Fleck, D. Han, Z. Qian, M. Heide, W. B. Huttner, P. Khaitovich, S. Paabo, B. Treutlein, and J. G. Camp. 2019. 'Organoid single-cell genomic atlas uncovers human- specific features of brain development', Nature, 574: 418-22.Schörnig, M., X. Ju, L. Fast, S. Ebert, A. Weigert, S. Kanton, T. Schaffer, N. Nadif Kasri, B. Treutlein, B. M. Peter, W. Hevers, and E. Taverna. 2021. 'Comparison of induced neurons reveals slower structural and functional maturation in humans than in apes', Elife, 10.
- Schörnig, M, Rava, V and Taverna, E(2023). Differentiation of rTTA/Ngn2-positive PSCs to iNeurons. Bio-protocol Preprint. 10.21769/p2308.
- Schörnig, M., Ju, X., Fast, L., Ebert, S., Weigert, A., Kanton, S., Schaffer, T., Nadif Kasri, N., Treutlein, B., Peter, B. M., Hevers, W. and Taverna, E.(2021). Comparison of induced neurons reveals slower structural and functional maturation in humans than in apes. eLife. DOI: 10.7554/eLife.59323
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.