
- Home
- Protocols

-

2.7. Docking computation
Last updated date: Nov 28, 2022 Views: 805 Forks: 0
Docking computation protocol for protein and its ligand
Detailed protocol:
This protocol enables the evaluation of protein (Native KIT, PDB ID: 4U0I)-ligand (Compound 06) interactions and binding properties to predict the activity of the ligand molecule.
Software and Module: Discovery Studio 2016 (DS 3.0), Discovery Studio Client (DS CDOCKER).
File types: 4u0i.dsv for protein, Compound 06.mol2 for ligand.
Protein preparation:
1. Native KIT (PDB ID: 4U0I) (4u0i.dsv) as a protein was used in this protocol, the 3D structure of protein native KIT (4U0I.pdb) was searched and downloaded from www.rcsb.org;
2. In Files Explorer, opened Samples/Tutorials/Receptor-Ligand Interactions, opened the protein file (4U0I.pdb) downloaded from PDB database (www.rcsb.org) (Figure 1);
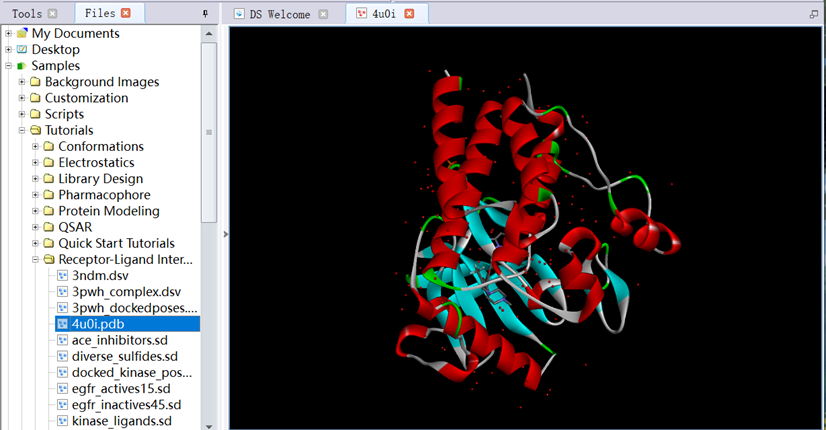
Figure 1 The three-dimensional structure of protein downloaded from PDB database
3. Ctrl+H, entered the System View. Ctrl+T, entered the Table View. In the System View, checked box of Water, clicked Cut to delete it;
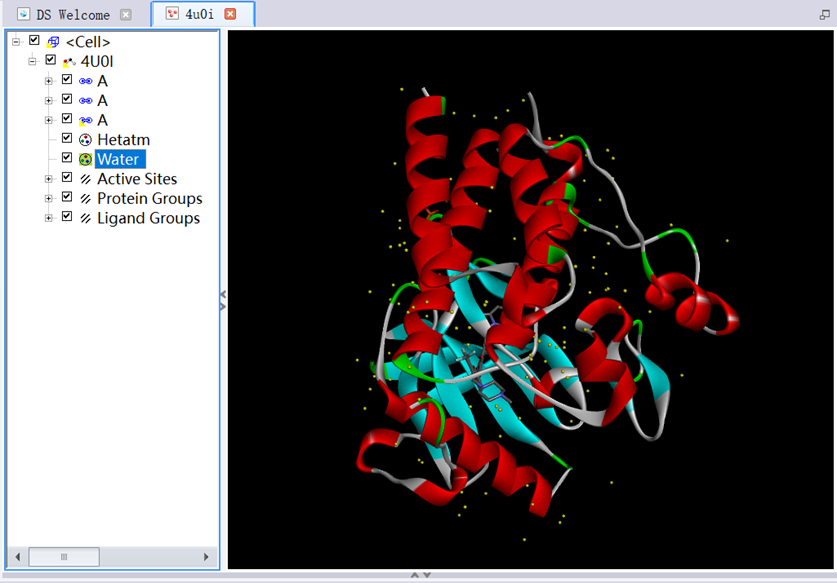
Figure 2 The three-dimensional structure of protein removed water molecules
4. In Tools Explorer, opened Macromolecules/Prepare Protein, clicked Clean Protein in Manual Preparation, clicked Prepare Protein in Automatic Preparation, clicked Run (Figure 3);
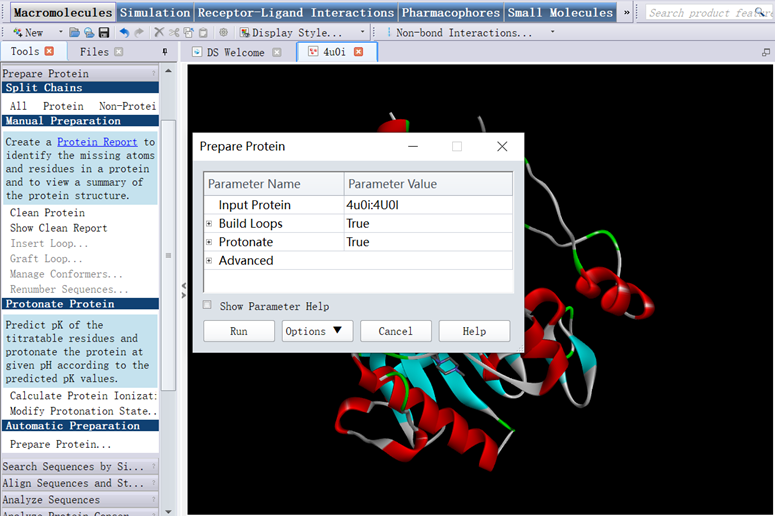
Figure 3 The three-dimensional structure of protein prepared for CDOCKER
5. In Tools Explorer, opened Receptor-Ligand Interactions/Define and Edit Binding Site/Define Receptor:4U0I, checked box of ligand Groups, Define Site/From Current Selection, checked box of ligand Groups clicked Cut to delete it saved as the protein file (4u0i.dsv) (Figure 4);
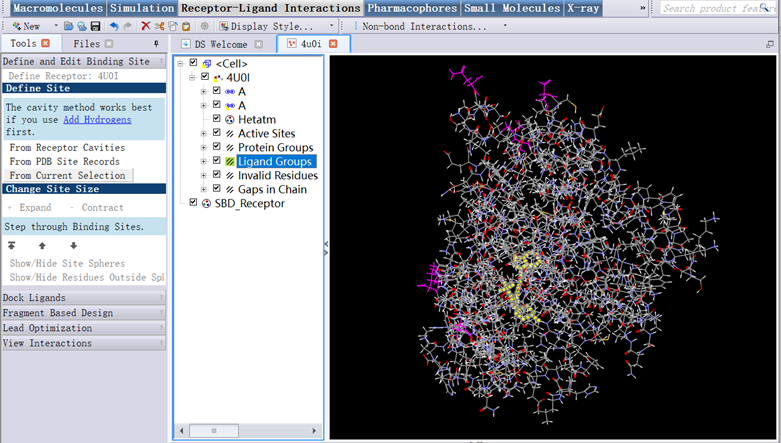
Figure 4 The three-dimensional structure of protein
Ligand preparation: Compound 06 (Compound 06.mol2) as a ligand was used in this protocol, the structure of ligand Compound 06 was prepared using ChemDraw 21.0.0 software (Figure 5).
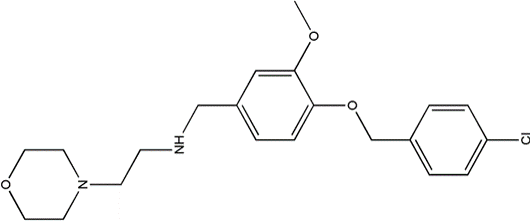
Figure 5 The structure of small molecule Compound 06
Preparation for CDOCKER:
1. In Files Explorer, opened Samples/Tutorials/Receptor-Ligand Interactions/4u0i.dsv (Figure 6);
Figure 6 The three-dimensional structure of protein
2. In Tools Explorer, opened Receptor-Ligand Interactions/Define and Edit Binding Site, clicked Show/Hide Residues Outside Sphere and Show/Hide Sphere, opened View/Transform, clicked Fit to Screen (Figure 7);
Figure 7 Amino acid in the active site of protein
3. In Files Explorer, opened Samples/Tutorials/Receptor-Ligand Interactions/ Compound 06.mol2 (Figure 8);
Figure 8 The structure of ligand
Running for CDOCKER:
1. In Tools Explorer, opened Receptor-Ligand Interactions/Dock Ligands, clicked Dock Ligands (CDOCKER);
2. Set parameters: Input Receptor (4u0i:4U0I), Input Ligands (Compound 06:All), Input Site Sphere (35.3194, 9.67323, 45.752, 12.5389);
3. Opened Top Hits, set parameters: Pose Cluster Radius (0.5), other parameters are shown in figure 9, including Random Conformations (10), Orientations to Refine (10), Simulated Annealing (True), then clicked run (Figure 9);
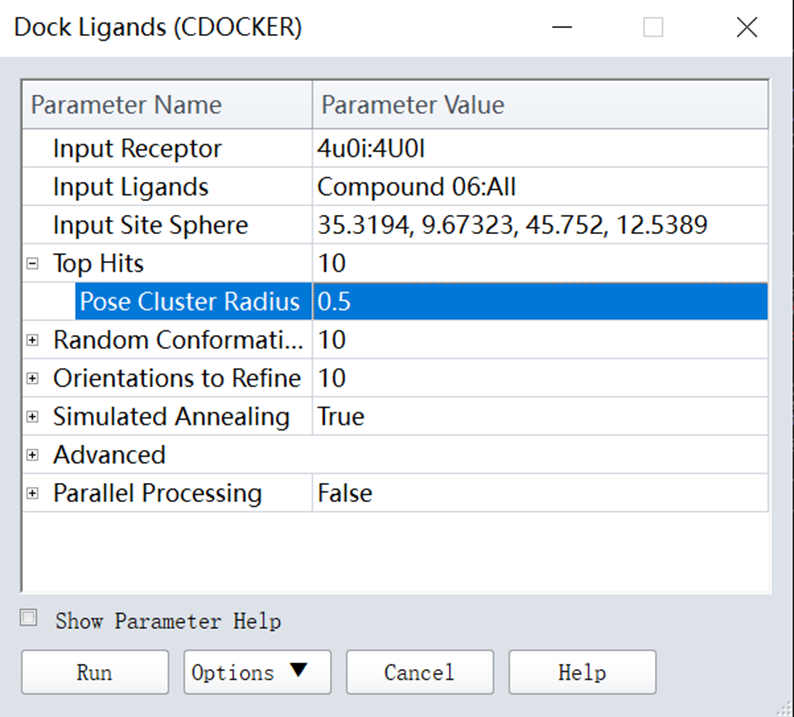
Figure 9 Parameters setting for CDOCKER
Results for CDOCKER:
1. In Tools Explorer, opened Receptor-Ligand Interactions/View Interactions, clicked Ligand Interactions (Figure 10);
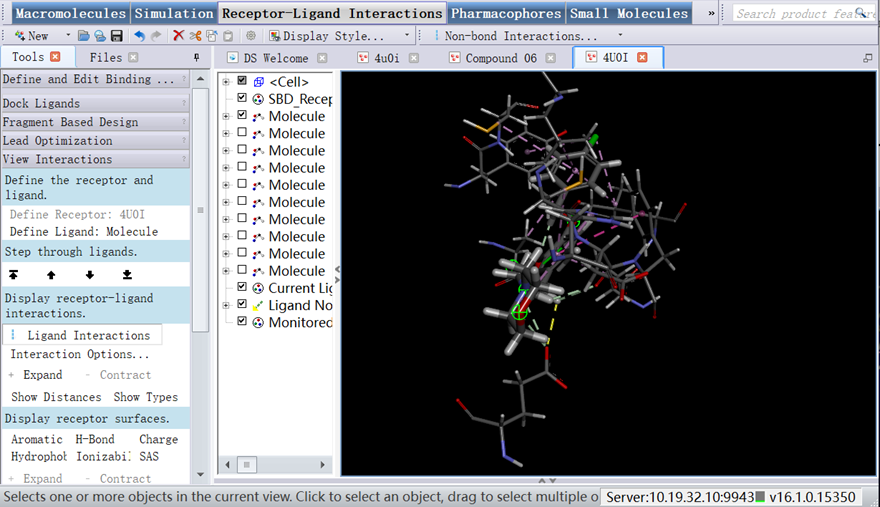
Figure 10 The interaction forces of protein-ligand
2. Clicked View Interaction/Interaction Options, as shown in figure 11;
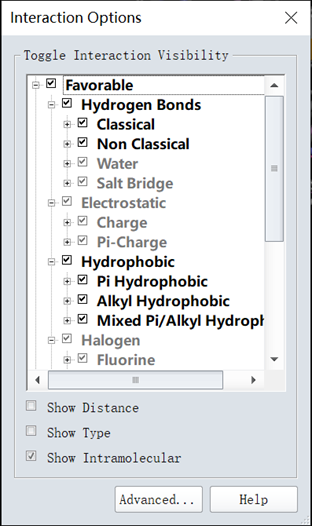
Figure 11 The interaction force types
3. Clicked View/Tool panels, checked the box of View Interaction (Advanced), selected the ligand you want to describe. In Tools Explorer, opened Receptor-Ligand Interactions/View Interactions, clicked Define Ligand, opened Receptor-Ligand Interactions/View Interactions (Advanced), clicked Show 2D Diagram (Two-dimensional diagram) (Figure 12);
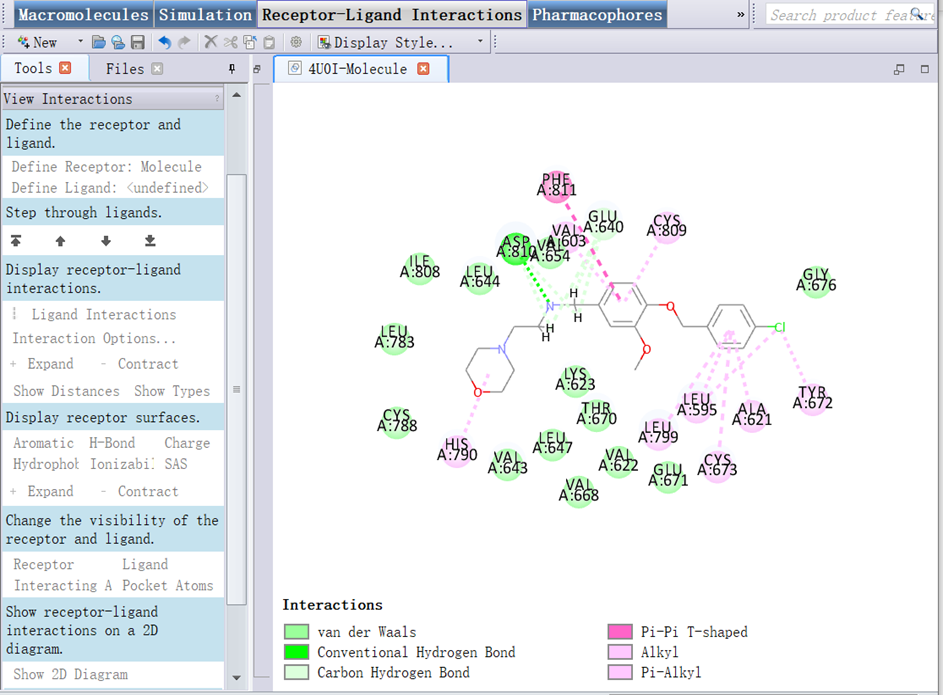
Figure 12 The 2D diagram interactions
- Jiang, L, Sun, J and Liu, Y(2022). 2.7. Docking computation. Bio-protocol Preprint. bio-protocol.org/prep2060.
- Jiang, L., Zhang, Z., Wang, Z. and Liu, Y.(2021). Discovery of novel potential KIT inhibitors for the treatment of gastrointestinal stromal tumor. Open Life Sci 16(1). DOI: 10.1515/biol-2021-0036
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.