
- Home
- Protocols

-

Immunoblot
Last updated date: Sep 14, 2022 Views: 824 Forks: 0
Dear Katja,
This is our latest protocol that was generated by Dr. Hiroyuki Mori from MacDougald lab.
He has the best Western blot skills in our lab. Hope this helps you.
Wish you the best luck!
Ziru & Ormond
Updated HM March 2022
Immunoblotting Protocol
“Life is a series of choices”
“Immunoblotting is a series of choices”
Preparation of lysate
1) Choose lysis buffer
| Lysis buffer | |
| Whole cell | NP-40 |
| Cytoplasmic (soluble) | Tris-HCl |
| Cytoplasmic (cytoskeletal bound) | Tris-Triton |
| Membrane bound | NP-40 or RIPA |
| Nuclear | RIPA or use nuclear fraction protocol |
| Mitochondria | RIPA or use mitochondrial fraction protocol |
https://www.abcam.com/protocols/sample-preparation-for-western-blot
Original lab protocol for lysis buffer
| SDS lysis buffer | ||
| Stock | vol mL | Final |
| 10% SDS | 10 | 1 % |
| 1 M TrisCl; pH 8.0 | 5 | 50 mM Tris-HCl |
| 0.5 M EDTA | 2 | 10 mM EDTA |
| H2O | 83 |
Alternatively, following buffers also work for cultured adipocytes and adipose tissue.
| NP-40 Mild lysis buffer | |||
| Stock | vol mL | Final | |
| 10% NP40 | 10 | 1 % | |
| 1 M TrisCl; pH 7.4 | 5 | 50 mM Tris-HCl | |
| 0.5 M NaF | 10 | 50 mM NaF | Target; Ser/Thr phosphatases |
| 0.5 M EDTA | 0.4 | 2 mM EDTA | Target; Metalloproteases (Mg2+, Mn2+) |
| PBS | 75 | ||
| TOTAL | 100 |
This is a popular buffer for studying proteins that are cytoplasmic or membrane-bound, or for whole cell extracts.
If there is concern that the protein of interest is not being completely extracted from insoluble material or aggregates, RIPA buffer may be more suitable as it contains ionic detergents that will more readily bring the proteins into solution.
| RIPA (radioimmunoprecipitation assay) | Final | |||
| Sodium chloride | 150 mM | |||
| NP-40 or Triton X-100 | 1.0% | |||
| Sodium deoxycholate | 0.5% | must be protected from light. | ||
| SDS (sodium dodecyl sulfate) | 0.1% | |||
| Tris, pH 8.0 | 50 mM | |||
| TOTAL | 100 | |||
2) Choose inhibitors
Protease and their inhibitors

Phosphatase and their inhibitors

The lab commonly uses “Protease Inhibitor Cocktail”
Sigma-Aldrich P8340-5ML in DMSO (Mammalian tissue) use 1:100 (kept in -20 oC)
Important: This product doesn’t contain EDTA nor phosphatase inhibitors (see the bottom table)
stocks of Na3VO4 and NaF are also in the -20 oC
| Components | |
| AEBSF | serine proteases |
| Aprotinin | Inhibits serine proteases such as trypsin, chymotrypsin, plasmin, trypsinogen, urokinase, and kallikrein |
| Bestatin | Inhibits aminopeptidases, such as leucine aminopeptidase proteases |
| E-64 | Inhibits cysteine proteases such as calpain, papain, and cathepsin B |
| Leupeptin | Inhibits both serine and cysteine proteases such as calpain, trypsin, papain, and cathepsin B |
| Pepstatin A | Inhibits acid proteases such as pepsin (human or procine), renin, cathepsin D, chymosin and protease B |
Alternatively, following individual inhibitors also work
For those who would like to use individual Inhibitor
| Inhibitors | uL for 10mL | Final | suggested | Target |
100mM PMSF in ethanol | 100 | 1mM | 0.1- 1.0 mM | Serine protease |
| Leupeptin 10mg/ml -20°C | 100 | 100 μg/ml | 1μM | Serine proteinases, plasmin, porcine kallikrein, and cysteine proteinases, endoproteinase |
Aprotinin liquid at 4°C | 2-10 μg/ml | Trypsin, chymotrypsin, kallikrein plasmin | ||
Pepstatin (in methanol) | 10 μg/ml | Aspartic acid proteases | ||
100mM Na3VO4 in water | 100 | 1mM | 1-100 mM | Tyrosine phosphatase |
Inhibitors can only slow down (not complete stop) the event including proteolysis, dephosphorylation and denaturation even samples are kept on ice.
For the study of protein acetylation
The main mechanism of protein acetylation is that acetyl donors (such as acetyl-CoA) transfer acetyl groups to the proteins under the catalysis of acetyltransferase
| Inhibitors | MW | Final | Target |
| Trichostatin A | 302.37 | 40 μM | Class I/II HDAC |
| EX-527 | 248.71 | 1 mM | SIRT |
| Nicotinamide | 122.12 | 400 mM | Class III HDAC |
| Sodium Butyrate | 110.09 | 200 mM | Class I/II HDAC |
3)-1 Preparation of lysate from frozen adipose tissue (also other tissues)
Make tissue powder using hammer
- Aluminum foil (about 15cmx10cm) is folded once, then fold again. In the middle, put adipose tissue and fold (as shown in the pictures).

- Put it into liquid nitrogen then put it on metal Heating Block upside down (flat side) with dry ice.
- Smash it using a hammer 4-5 times. Extreme low temperature is important to make tissue powder, so place back in liquid nitrogen to chill down, then repeat smashing.
- This powder is possible to use for protein or RNA Extraction.
- After making powder, it is possible to store at -80oC until further processing.
Note: this method works for any tissue including muscle, skin etc.

For homogenization
- Pellet Pestle Motor is used .
- Put adipose tissue powder in the Eppendorf tube up to ~ 100µL line of a tube, then add 200-300 µL of lysis buffer.
- Homogenize for 10-20 seconds on ice.

- About 15 min after homogenization, centrifuge lysates at 20,000 x g for 15 min at 4oC.
- Remove the top lipid layer, or you can make a hole in the lipid layer, then penetrate the hole with a pipette to get water layer. Transfer the extract to a new tube that is prechilled on ice.
- Centrifuge extracts again to get cleaner lysate.
- Transfer to a newtube for protein assay.
3)-2 Preparation of lysate from cultured adipocytes
Preparation of lysate from tissues
- If using the following volume of lysis buffer, you get the lysate whose concentration is around
1.0-1.5 µg/µL (depending on cell confluency, differentiation status).
100-130 µL of lysis buffer for one of 12 well plates,
200-250 µL of lysis buffer for one of 6 well plates.
- Wash the cells with ice-cold PBS (directly to plates)
- Aspirate the PBS, then add ice-cold lysis buffer.
- On ice, scrape and mix cells with lysis buffer using a cell scraper or backside of P1000 tip.
- Transfer the lysate into a pre-cooled microcentrifuge tube.
- About 15 min after homogenization, centrifuge lysates at 20,000 x g for 15 min at 4oC.
- Following step is same as tissue processing above.
4) Protein assay
| assay | Abs. (nm) | mechanism | detection limit (µg/mL) | advantages | disadvantages |
| UV absorption | 280 | tyrosine and tryptophan absorption | 0.1-100 | small sample volume, rapid, low cost | incompatible with detergents and denaturating agents, high variability |
| Bicinchoninic acid | 562 | copper reduction (Cu2+ to Cu1+), BCA reaction with Cu1+ | 20-2000 | compatible with detergents and denaturing agents, low variability | low or no compatibility with reducing agents |
| Bradford | 470 | Complex formation between Coomassie blue and proteins | 20-2000 | compatible with reducing agents, rapid | incompatible with detergents |
| Lowry | 750 | copper reduction by proteins, Folin-Ciocalteu reduction by the copper-protein complex | 10-1000 | high sensitivity and precision | incompatible with detergents and reducing agents, long procedure |
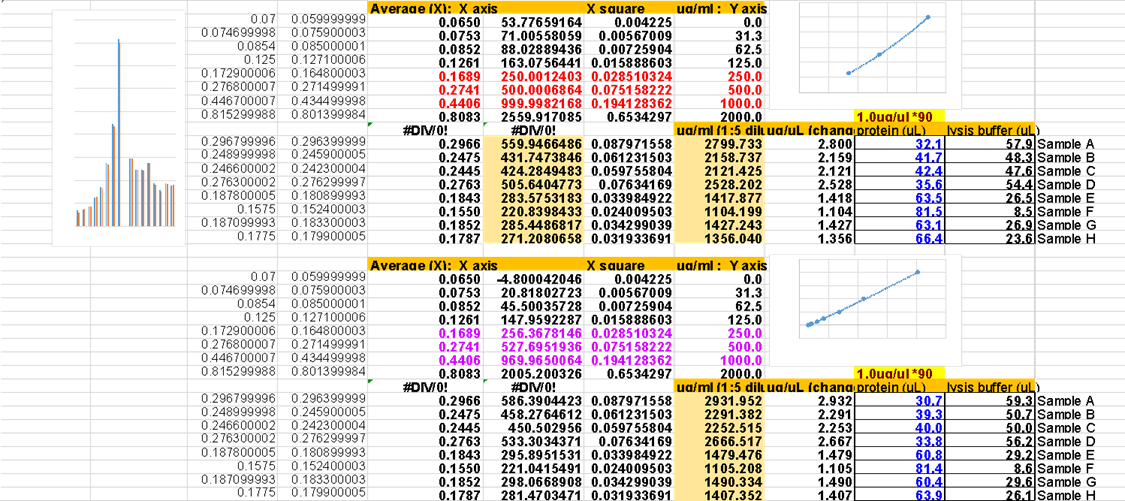
Our lab uses BCA protein assay (Thermo Scientific Pierce, PI23235).
- Preparation of diluted Albumin (BSA) Standards (aliquot to 8 lane tubes can be stored at -20 oC)
A 2.0 mg/mL of BSA original vial from the kit
B 1.0 mg/mL
C 0.5 mg/mL
D 0.25 mg/mL
E 0.125 mg/mL
F 0.0625 mg/mL
G 0.03125 mg/mL
H water
- In order to start the reaction at the same time (there is a time lag for pipetting), 96 wells microplate well is put on ice.
- To prevent plate scratching by ice, a paper towel is put between ice and microplate.
- Pipette 10 µL of standard replicate into a 96 wells microplate
- Pipette 8 µL of water into a 96 wells microplate well for dilution of the unknown sample .
- Pipette 2µL of unknown sample into water in 96 wells microplate (samples are diluted 1:5).
- Prepare BCA Working Solution (100 µL/well)
Around 10 mL for 1 plate; Mix 10mL of Reagent A with 200 µL of Reagent B (Reagent A:B=50:1)
- Add 100 µL of BCA Working Solution to each well, then. Incubate 30 minutes at 37°C.
- Measure absorbance at 562 nm. (plate can be reused a few times after cleaning)
Wavelengths from 540-590nm have been used successfully with this method.
Compatible substance concentrations (inhibitor, detergent, etc) are available as follows
https://assets.fishersci.com/TFS-Assets/LSG/manuals/MAN0011237_Micro_BCA_Protein_Asy_UG.pdf
- Dilute lysates to equal protein concentrations in lysis buffer and add 4x Laemmli sample buffer.
- Samples are heated at 95°C for 5 min, then immediately cooled down on ice.
(keep samples at -20°C for future use)
- Load <20 µg protein per well (usually 5-15µg) (before adding Laemmli sample buffer) is enough for immunoblotting.
- Higher amounts of lysate per well cause the proteins to migrate inconsistently (inverse triangular shape).
Laemmli 4X buffer / loading buffer | Amount for 25mL | Final |
| SDS | 2 g | 8% SDS |
| 1M Tris-HCl pH 6.8 | 5 mL | 200m M |
| glycerol | 10 mL | 40% glycerol |
| 2-mercaptoethanol | 5 mL | 20% |
| (1M DTT) | 1 mL | 50mM (replacement for 2-mercaptoethanol) |
| 0.4% bromophenol blue | 0.1 g | 0.4% bromophenol blue |
| Total | 25mL by water |
Preparation of lysate 5) Choose gels
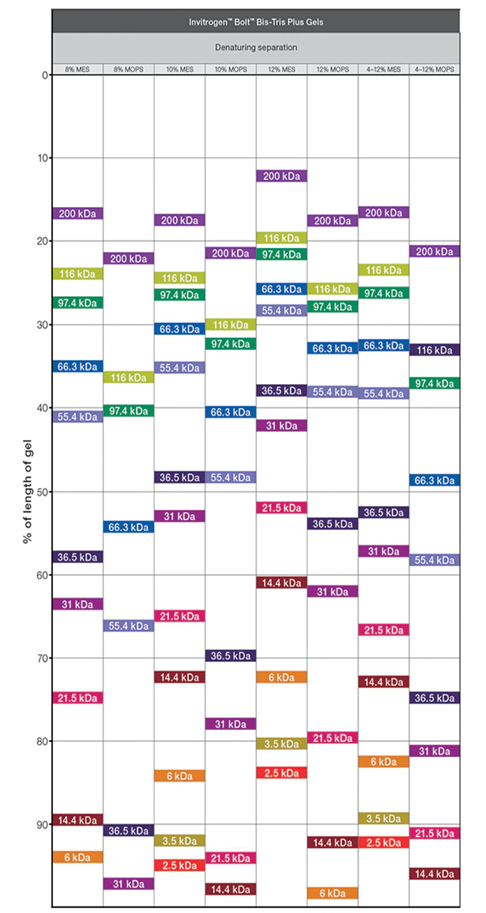
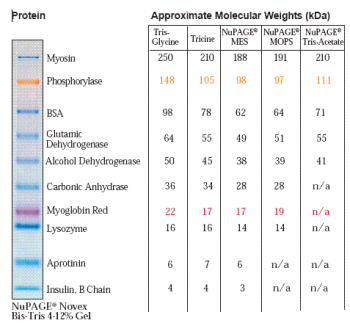
Although we use Bis-Tris midi gels (almost no smiling) 4-12% (for broad range separation), there are many choices depending on which sizes of proteins you are interested in. When ordering, we need to choose “w/adapters” since our gel tank is from BioRad not from Invitrogen
WG1403A: NuPAGE™ 4-12% Bis-Tris Midi Protein Gels, 26-well, w/adapters
WG1402A: NuPAGE™ 4-12% Bis-Tris Midi Protein Gels, 20-well, w/adapters
Following is the “Protein Gel Selection Guide” in ThermoFisher web
Although we normally use MOPS buffer, MES buffer can focus smaller protein even using same gels.
MOPS buffer: 30 to 260 kDa
MES buffer: 2.5 to 260 kDa
Following is the migration charts from ThermoFisher web.
SDS page, transfer
5) Loading and running the gel
MOPS SDS Running Buffer
- To prepare 1000 mL of 10X MOPS SDS Running Buffer, dissolve the following reagents to 800 mL ultrapure water (from original NuPage protocol)
MOPS 104.6 g, Tris Base 60.6 g, SDS 10 g, EDTA 3.0 g
- Mix well and adjust the volume to 1000 mL with ultrapure water.
- For electrophoresis, dilute this buffer to 1X with water.
The pH of the 1X solution is 7.7.
- Lab protocol: 5X MOPS Running Buffer 3.5L
MOPS 183.1 g, Tris Base 106.1g, SDS 17.5 g, EDTA 5.25 g
20X MES SDS Running Buffer Catalog number: B0002
- Load <20 µg protein per well (usually 5-15µg) (before adding Laemmli sample buffer) is enough for immunoblotting.
- Load protein marker (SeeBlue™ Pre-stained Protein Standard: LC5625); 4-5 µL is enough.
Load a marker into both side of the wells, you could cut a membrane to small piece based on MW.
- Based on the NuPAGE Gels information, it is suggested to run the gel at the 200 V constant.
However, this is too high especially at the stacking area of the gels.
- If too high voltage for loaded proteins, dye shape become triangle as below
(left: normal, right: too high voltage)

- Start: 30V, then increase to 60-100V after the top dye reach to 1/5-1/4 of gel, then keep until the samples reach the middle of the gel
- Finally, run at 120-180V until the end of running.
- Make sure whether the top of the running buffer is enough or not every 30 min since we use Invitrogen gels with BioRad tank with adaptor.
- Even tightly attach the adaptor to the gel, the running buffer in the top leaks slightly, which causes the stop running or running uneven due to the electrodes line is not covered with ruining buffer.
- Stop running when a marker protein that is close MW with a protein you are interested in migrates an adequate area of a gel.
6) Transferring the protein from the gel to the membrane
| 10x Transfer buffer | Amount for 16 L | Final (1x) |
| Tris base | 484 g | 25 mM |
| glycine | 2304 g | 190 mM |
| SDS | 8 g | 0.05 % |
| Total | 16 L by MilliQ water |
1x Transfer Buffer (1L)
100 ml 10x Transfer buffer
200 ml MeOH
700 ml MilliQ water
PVDF membrane: (Immobilon-P PVDF Membrane, EMD Millipore IPVH00010)
- Pre-wet PVDF membrane for 30 seconds in methanol, then pour extra methanol into transfer chamber. Briefly rinse in deionized water, then place it in Transfer Buffer for several minutes.
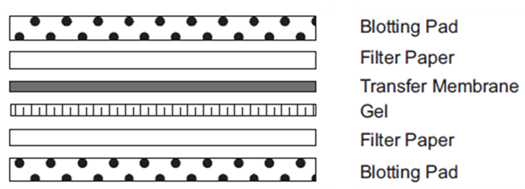
- Prepare the stack as follows: (set black side of holder down first? and red on top)
In general, one filter paper of each side is enough. However, the blotting pad is older and being thin,
it is better to adjust the thickness (1 of each side => 1 for bottom and 2 for top => 2 of each)
Too much tight stack causes gel cracking during the transfer.
- The PVDF membrane we use is rolled. If you plan to incubate over 2 membranes into the same box for primary-secondary Abs, it is better to transfer inside the roll (see the middle and right of figures below), because membranes stack to the box during antibody incubation if transferring outside of the roll (left figure).

- In order to decrease methanol waste for the environment, re-use transfer buffer is recommended.
e.g. half new and half re-used one.
- Perform the transfer at 700 mA constant for 120-150 minutes for midi-gels.
- Always check the voltage at the starting point in order to confirm you use the correct buffer.
The expected Current;
At the start is around 160 V (all new buffer) and 120 -130 V (half new and half reused).
At the end of transfer, it becomes 70-80 V.
- Pause the power supply whether air bubbles come black electrodes (+) or not. If air bubbles come to the red side you might connect electrodes the opposite way.
- Change the ice pack inside the transfer tank every 30 min in order to keep the transfer buffer cold.
Optional; cover the transfer tank with ice.
Primary Ab, secondary Ab, and detection
7) Blocking- primary antibody
| 10x TBST like buffer (wash buffer) | Amount for 1 L |
| NaCl | 200 g |
| 1M TrisCl pH 8.0 | 20 mL |
| Tween 20 | 5 mL |
| Total | 1 L |
This buffer is not exact TBST; the original idea was from “Jikken-igaku”, a Japanese research
magazine, that collected ideas for fuss-free experiments from readers in some duration as mini-issue.
The concept of the mini-issue was how easy to make buffers using solutions/buffers most labs have.
Blocking buffer: 5% nonfat dry milk (grocery store grade)in the wash buffer
Primary antibody buffer: 5% BSA in the wash buffer with Sodium azide (final 0.02% (w/v))
- After finishing transfer, remove a filter paper on the gel, then cut a membrane based on the marker
after putting a ruler on the gel (transferred proteins on the membrane are protected by gel).
- Incubate membrane with blocking buffer for 30-60 min at room temperature
- Rinse twice by the wash buffer to get rid of milk.
- Incubate the membrane with primary antibody solution (always fix 1:1000; the intensity of
Immunoblot-bands is controlled by secondary Ab concentration) overnight at 4°C.
8) Secondary antibody, detection, reblot
- Collect primary antibody to 15 mL of polystyrene tube for re-use, then keep at -20°C (Not polypropylene)
- Rinse twice with the wash buffer, then wash with the wash buffer every 10 minutes for 30 min.
- Incubate the membrane with the 2ndary antibody in the blocking buffer at room temperature for 1 h.
(Newly used antibody (have no experience to use it), try to start 1:3,000 dilution), then adjust
secondary antibody concentration based on the experience of 1st trial)
- Rinse twice by the wash buffer, then wash with wash buffer every 10 minutes for 30 min
- Rinse with the wash buffer without Tween 20 (Tween 20 weaken ECL signal in some cases)
- We have 3 different ECL; the sensitivity <<< price<<<
- After finishing detection, wash off ECL relatively quickly after imaging to prevent bands from "burning" into membrane
| 10x Stripping buffer | Amount for 1 L | Final (1x) |
| Glycine | 18.8 g | 25 mM |
| SDS | 100 g | 1% |
| Total | 1 L | pH 2.0 |
Re-pH 2.0 after making 1x
- Strip membrane by stripping buffer for 20 min (change to new buffer 3 times during 20 min).
- Rinse with DI water for completely get rid of stripping buffer.
- Following steps including blocking are same as fresh membrane.
Protocol for analysis of secreted adiponectin multimers (From Will CAWTHORN)
Reagents needed
________________________________________________________________________
Non-reducing, partially denaturing sample buffer (1X)
3% SDS
50 mM Tris-HCl pH 6.8
10 % glycerol
0.05 % Bromophenol blue
________________________________________________________________________
MOPS SDS-PAGE running buffer (5X): Same as our MOPS running buffer
________________________________________________________________________
Western blot transfer buffer: Same as our transfer buffer
________________________________________________________________________
Western blot washing buffer (10X): Same as our wash buffer
________________________________________________________________________
Adiponectin primary antibody
Company: Sigma
Cat #: A6354-200UL
Species: Rabbit
URL: http://www.sigmaaldrich.com/catalog/product/sigma/a6354?lang=en®ion=US
______________________________________________________________________
Protocol: non-reducing, partially denaturing conditions
Mix 1.5 µL of serum with 24 µL non-reducing, partially denaturing sample buffer; any dilution of serum from 20- to 50-fold in this loading buffer seems to work. Once mixed, do not heat-denature, but incubate at room temperature for approx. 20 min. Doing this seems to allow the multimers to be better resolved on the gels.
Load on gradient gels (e.g. Invitrogen Novex Midi Bis-Tris gel, 4-12%; or BioRad Criterion Midi Tris-HCl gel, 4-15%) and run samples in MOPS SDS-PAGE running buffer (made to 1X with distilled water). Using this approach, the low-molecular-weight adiponectin multimers run at around 67 kDa, with the high-molecular-weight multimers running above 150 kDa and the middle-molecular-weight multimers running just below the HMW multimers. It is a good idea to run the samples for long enough to adequately separate the different multimers (e.g. until the ~30 kDa marker bands are near the bottom of the gel).
Transfer proteins to PVDF membrane using 1X Western blot transfer buffer.
Block, incubate in primary and secondary antibodies, and expose according to standard western blotting procedures.
Based on the experience following incubation time may be adequate for culture cells.
For 3T3-L1s: 4-hour incubation,
MSC adipocytes 4 hours incubation
Rat primary adipocytes 75 minutes
Intact adipose tissue explants: 4-hour incubation
- Li, Z and MacDougald, O A(2022). Immunoblot. Bio-protocol Preprint. bio-protocol.org/prep1942.
- Lipolysis of bone marrow adipocytes is required to fuel bone and the marrow niche during energy deficits. eLife. DOI: 10.7554/eLife.78496
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.