
- Home
- Protocols

-

Protocol of full article
Last updated date: Feb 23, 2022 Views: 775 Forks: 0
Protocol for “Annotating Putative D. discoideum Proteins Using I-TASSER” Ryan Rahman
Texas A&M University
The objective of this protocol is to provide a tool (one of many) that can be implemented to characterize the functions of poorly understood gene products that have no gene ontology (GO) annotation. One such method implemented by the lab of Dr. Richard Gomer is inputting protein FASTA sequences into a comparative suite of bioinformatic programs called I-TASSER, which was developed and is currently maintained by the Zhang lab at the University of Michigan.
Important Links:
- NCBI BLASTp, UniProt, PFAM, InterPro,STRING,ProtScaleExpasy
- I-TASSER server for protein structure and function prediction (zhanggroup.org)
- What is I-TASSER? C-score TM-score
Stepwise protocol:
Identifying candidate genes/gene products
4. A prerequisite for I-TASSER is the assumption that the genome of the model organism of choice has been sequenced and these data are readily available. As an example, I will illustrate the steps for Dictyostelium discoideum.
5. Navigate to the model organism database (Dictybase.org for D. discoideum) and identify unannotated genes (either those of interest obtained through proteomic, transcriptomic, or genomic data or by contacting the curators of said database).
- Search for the gene product of interest utilizing a gene ID or sequence search. Obtain the FASTA protein sequence for the gene of interest.
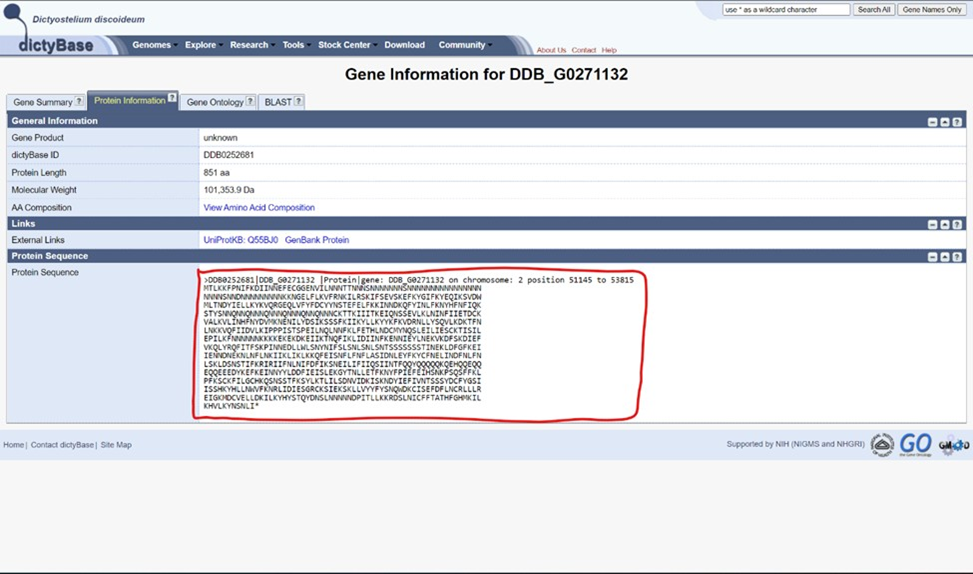
b. Verify that the gene of interest is unannotated on the database (DictyBase) and perform a BLASTp (protein-to-protein) search. BLAST, or Basic Local Alignment Search Tool, compares the inputted sequence to all those stored by NCBI to find those that share the most sequence similarity (read more about the exact details of how BLAST works). If there is a great hit with high similarity, then it will have a very small e value (which is similar to a p-value in that the lower it is, the more statistically significant the result is). If there is a hit that is well-characterized in another organism, then an annotation can already be written based on the functional information about the other gene product, which is essentially an ortholog of your protein. If there is a low e value hit, or multiple hits, but all the hits are ‘hypothetical protein’ then what you are looking at is a protein of unknown function in Dicty that does need to be investigated using I-TASSER. Here is the link to NCBI BLASTp.
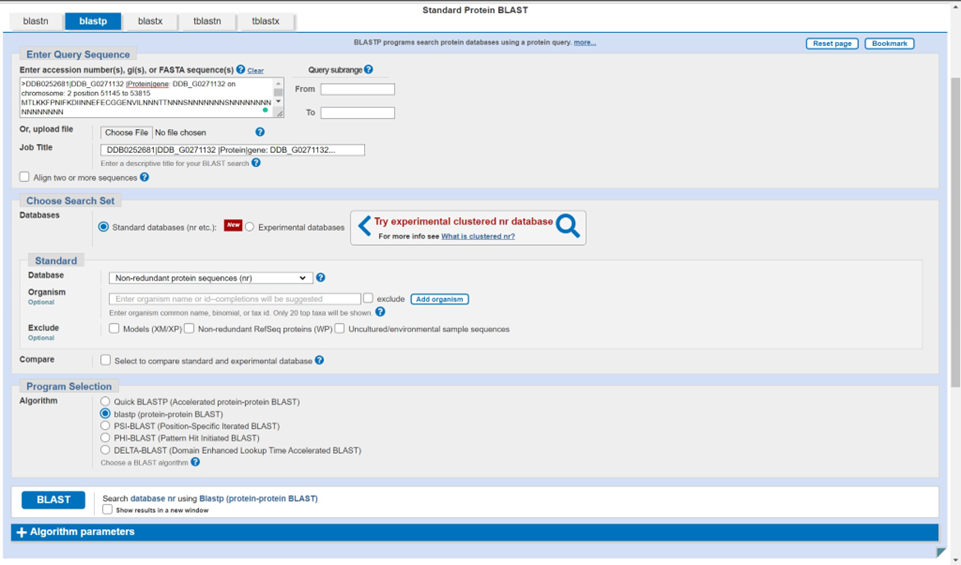
6. Include your top 5 BLASThits in the final annotation of your gene/gene product (if the e valueis below the widely accepted threshold value of 0.01). If BLAST yields very few results, or just hypothetical proteins, or more information is desired, proceed with I-TASSER using the following website: I-TASSER server for protein structure and function prediction
(zhanggroup.org).
- TASSER gene annotation
7. To utilize this resource, registration is required and can take a couple of days to process (it is free of charge, however as long as you register with a .edu email address). After receiving the confirmation email and password for your account, login on the home page to submit the sequence for your protein of interest.
- After clicking “Run I-TASSER” the suite of programs will run on the University of Michigan supercomputer cluster. Depending on the time of year, this can take anywhere from a few days to several weeks. While waiting for the results, you can run other bioinformatic analyses to determine if the gene product has any hits on UniProt, PFAM, InterPro, STRING, etc. and construct hydrophobicity plots (typically we use Kyle and Doolittle plots). These are all tools that can be used with your single FASTA protein sequence.
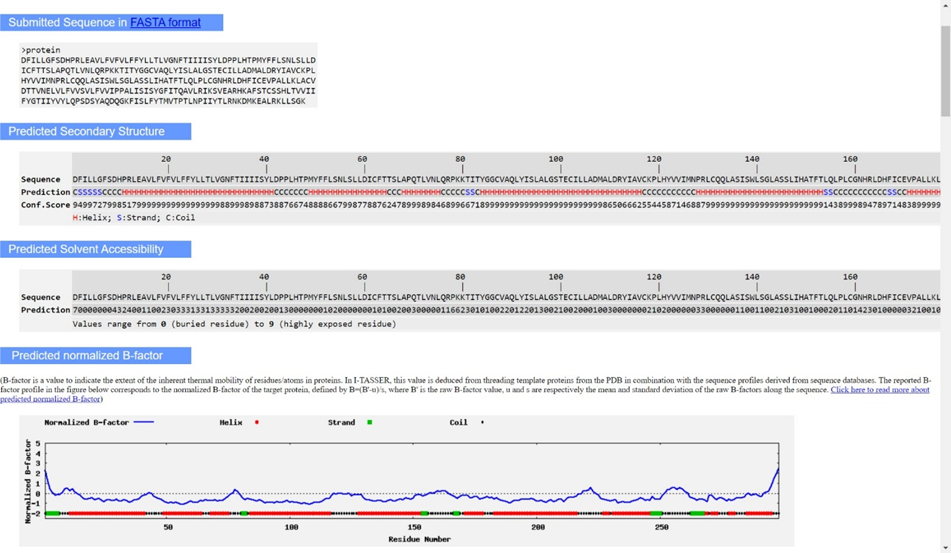
- Once you receive the output of an I-TASSER job, it can be difficult to interpret the results. It is important to understand the basic concepts behind how the program produces an output before analyzing the results. First, I-TASSER runs some typical sequence- based predictions of secondary structure and hydrophobicity based on the properties of each residue. Next, I-TASSER takes the given sequence and uses Protein Data Bank (PDB) x-ray crystallography structures to construct multiple threading templates through LOMETS (a unique meta-threading approach). If LOMETS cannot find any great matches in PDB, it implements ab initio modeling. The templates are then used to create several three dimensional models based on probabilistic computations concerning structure similarity, not sequence similarity. Through SPICKER clusters, the alignment and assembly of these threads is assessed based on spatial constraints (steric clash is minimized and low free energy is maximized). The top ten threading templates are then outputted based on a “Z-score” that measures the performance of the template; a bigger z- score means a greater deviation from the average performance of other templates (which is desired because we want the best templates). These templates are NOT, however, always the final proteins that are most similar to the gene product of interest. Those come further down in the output.
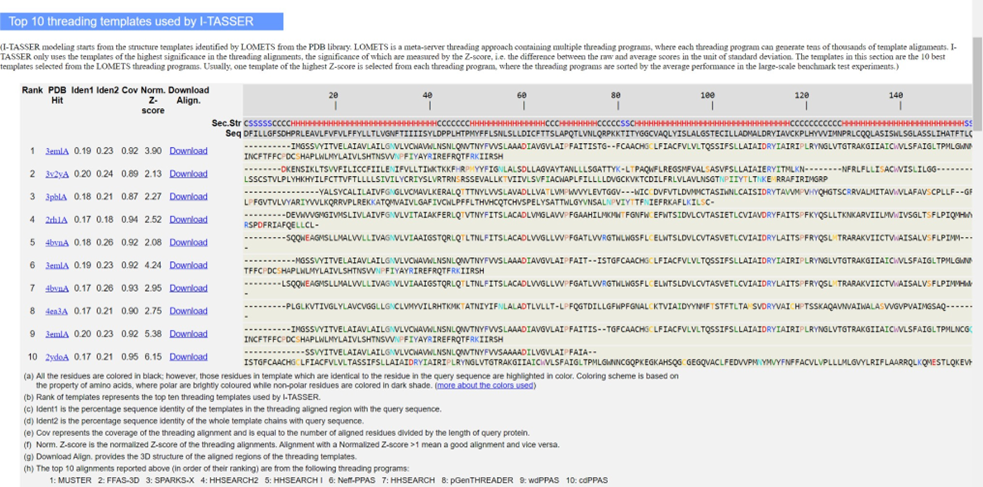
c. With the use of the best threading templates, several SPICKER cluster simulations are run to create the top 5 three-dimensional models which are then given C-scores. “C-score is a confidence score for estimating the quality of predicted models by I-TASSER. It is calculated based on the significance of threading template alignments and the convergence parameters of the structure assembly simulations. C-score is typically in the range of [-5,2], where a C-score of higher value signifies a model with a high confidence and vice-versa.”

d. The most informative and functional data given by I-TASSER are displayed in the following sections of the example output: “Proteins structurally close to the target in the PDB” and “Predicted function using COFACTOR and COACH”.
i. The first subsection illustrates the best final I-TASSER model (“Model 1” from above), and I-TASSER compares its structure to all x-ray crystallography structures in the PDB using TM-Align. These are the proteins with the most structural similarity to your gene product of interest! Therefore, the proteins with the highest TM-score are most likely to share the same function with your gene product of interest since it is mostly accepted that form follows function, especially when it comes to proteins. Click on the hyperlinks to the PDB hits and note the predicted functions and protein families/classes of the top five hits assuming these hits have a TM-score > 0.50 (the accepted cutoff for TM- scores).

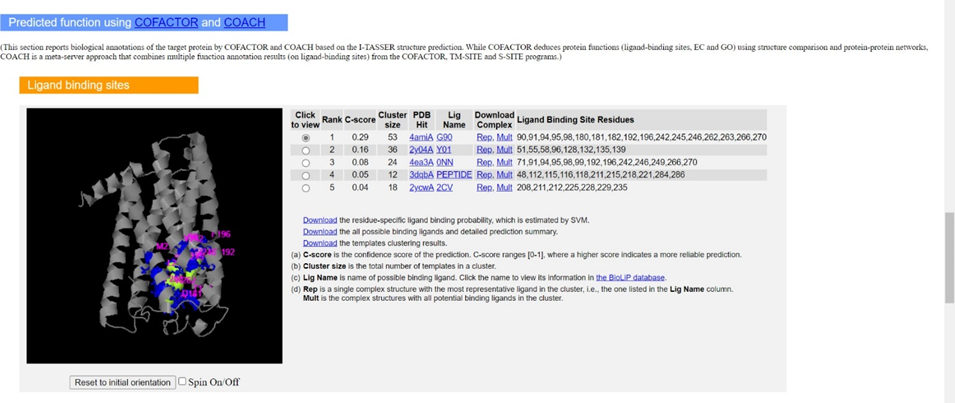
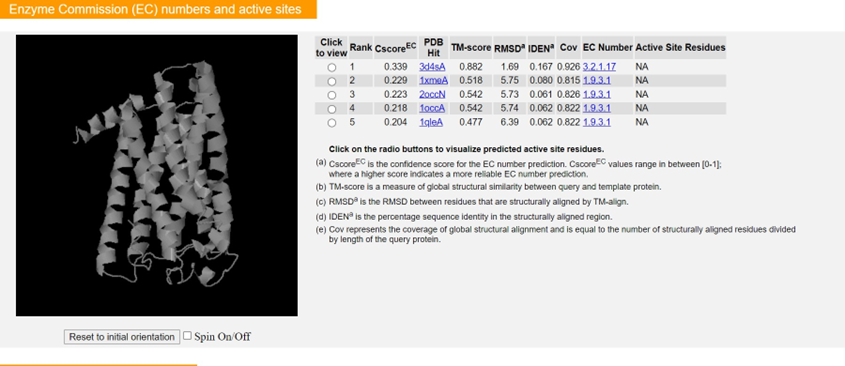
ii. The final subsections include predictions of motifs such as ligand-binding sites, catalytic/active sites, and the gene ontology of your gene product of interest. Again, utilize the C-scores and GO-scores to assess which hits are accurate for your gene product. Information concerning the calculation of these statistical measures can be found in the Zhang lab’s 2015 Nature Methods paper, and a concise annotation of the example output is also available here.If you prefer an even more technical protocol about how to use I-TASSER, use this link.
e. Lastly, please remember to cite the Zhang lab in all publications associated with I- TASSER:
i. You are requested to cite following articles when you use the I-TASSER server:
- J Yang, R Yan, A Roy, D Xu, J Poisson, Y Zhang. The I-TASSER Suite:
Protein structure and function prediction. Nature Methods, 12: 7-8 (2015). (Download the PDF file). - A Roy, A Kucukural, Y Zhang. I-TASSER: a unified platform for automated protein structure and function prediction. Nature Protocols, 5: 725-738 (2010) (download the PDF file)
- Y Zhang. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics, vol 9, 40 (2008). (download the PDF file).
Final Annotations
- After collecting all data, write a succinct annotation listing every C-score, TM-score, GO-score, and e value from every tool. The Gomer lab used the following format guide and statistical cutoffs:
Protein ID
- Top 5 I-TASSER hits, and name/ function of the hit (with TM-score):
- GO terms for the I-TASSER hits:
- Protein BLAST hits (blast predicted amino acid sequence against all known proteins) (give w/super low E value, name and function of the BLAST hit):
- Co-expression information (give the name/ function of the protein where there is co- expression of the mRNA from STRING db)
- Hydrophobicity: Kyte/Doolittle plot from ExPasy
- Conserved domains and motifs: (PFAM and InterPro)
- Shape: (globular, fibrous, etc)
- Binding activity/sites: (metal binding, DNA-binding, ATP-binding, etc)
- 3D structures
- Predicted activity/cellular localization: (nuclear, receptor, transport channel, etc)
- Any other pertinent info from other sources
Statistical cutoffs for I-TASSER
- Threading templates- According to I-TASSER, a norm. Z-score >1 is GOOD for the templates, because the normalization is calculated by dividing the Z score by a target value. From the I-TASSER publication: “However, since LOMETS contains templates from multiple threading programs where the Z-scores are not comparable between different programs, I-TASSER uses a normalized Z-score (highlighted by the orange box) to specify the quality of the template, which is defined as the Z-score divided by the program-specific Z-score cutoffs. Thus, a normalized Z-score >1 indicates an alignment with high confidence.”
- Models- C-score is calculated by the following formula: “The confidence of each structure model is estimated by the confidence score (C-score), that is defined by Equation 1: C−score=ln(M/Mtot⟨RMSD⟩∗1N∑i=1NZiZcut,i) whereM/Mtot is the number of structure decoys in the SPICKER cluster divided by the total number of decoys generated during the I-TASSER simulations. 〈RMSD〉 is the average RMSD of the decoys to the cluster centroid. Zi/Zcut,i is the normalized Z -score of the best template gene, rated by the ith LOMETS threading program. Our large-scale benchmark tests showed that the C-score defined in Equation1 is highly correlated with the quality of the predicted models (with a Pearson correlation coefficient >0.9 to the TM-score relative to the native) (Zhang, 2008). The C-score is normally in [−5, 2] and a model of C-score >−1.5 usually has a correct fold, with TM-score >0.5. Here, TM-score is a sequence length-independent metric for measuring structure similarity with a value in the range [0, 1]. A TM score >0.5 generally corresponds to similar structures in the same SCOP/CATH fold family” (Xu and Zhang, 2010).
2. An example is provided here as a bulleted list for DDB_G0271132:
DDB_G0271132
Annotation:
- BLASTp yields a lot of other hypothetical proteins, but none that are greater than 50% similar. Most don’t have a description except for a couple cell surface proteins, but their e-values are high. cell surface protein DTFA [Dictyostelium discoideum AX4] (e-value = 0.056).
- UniProtKB: describes two coiled-coil motifs.
- STRING: Only one gene coexpression on STRING with another hypothetical Dictyostelium gene product.
- PFAM/InterPro: Coiled-coil motif
- I-TASSER results: Top 3D model has a C-score of –1.21 on a scale of [-5,2], and has a TM-score (statistical structural alignment from [0-1] where ~0.5 andabove indicates a correct topology) of 0.56 +/- 0.15.
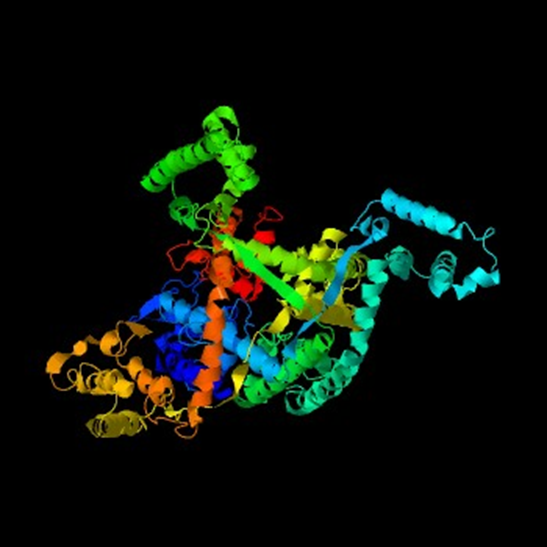
○ Predicted secondary structure: 35-36 separate alpha helices, 7-8 beta strands, and the rest of the residues are associated with coils.
○ Most of the amino acid residues are buried, implying that they are likely hydrophobic and could be components of a transmembrane channel.
○ I-TASSER uses LOMETS to access the PDB library and perform complex multithreading to compare tens of thousands of template alignments to the query sequence. The top 5 threading templates are given by the codes: 6kzoA, 5fvmA, 6r9tA, 1vt4, and 1vt4A. These are listed in descending rank, with normalized Z-scores of 1.50, 1.68, 1.39, 1.38, and 1.17 respectively. Coverage values (calculated as number of structurally aligned residues divided by length of query) for each hit were found to be 0.94, 0.97, 0.98, 0.61, 0.17.
○ The top 5 protein structural analogs were identified by the PDB database by the codes 6kzoA, 6uz0A, 3ir7A, 1sijA, 1dgjA, which corresponds to a human voltage-gated calcium channel (membrane protein), a sodium channel (membrane protein) for cardiac action potentials, a transmembrane oxidoreductase, an oxidoreductase (aldehyde dehydrogenase), and an oxidoreductase, respectively.
○ The top 4 ligand binding sites are given by the ligand names MG, FES, ZN, and MG which correspond to magnesium, and iron-sulfur cluster, zinc, and magnesium with C-scores of 0.07, 0.02, 0.02, and 0.02 respectively.
○ The top 5 enzyme commission (active sites) PDB hits were given by the codes 2j5wA, 1g8kA, 1kgfA, 1dgjA, and 1h0hA with TM-scores of 0.270, 0.261, 0.253, 0.277, and 0.247 respectively in descending rank. These correspond to
a human metal-cation binding oxidoreductase, an argenite oxidase, a dehydrogenase, an aldehyde oxidoreductase, and a formate dehydrogenase electron transport protein respectively.
- Consensus prediction of gene ontology by I-TASSER is as follows: (Parenthetical GO-Score given in the range of [0,1] based on a weighted measure C-score(GO).)
- Molecular function predictions: GO:0051540 (0.56), GO:0016491 (0.35), GO:0046914 (0.31) which correspond to metal cluster binding, oxidoreductase activity, and transition metal ion binding respectively.
- Biological process predictions: GO:0051234 (0.41), GO:0055114 (0.35) which correspond to establishment of localization and oxidation-reduction process.
- Cellular component: none was predicted.
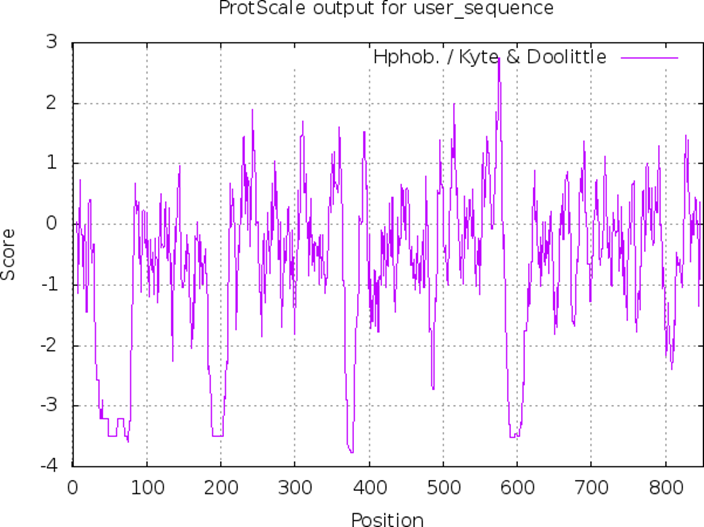
- Kyte and Doolittle Hydrophobicity Plot:
- The Kyte-Doolittle scale is widely used for detecting hydrophobic regions in proteins. Regions with a positive value are hydrophobic.
- Rahman, R and Gomer, R H(2022). Protocol of full article. Bio-protocol Preprint. bio-protocol.org/prep1545.
- Annotating Putative D. discoideum Proteins Using I-TASSER. microPublication Biology. DOI: 10.17912/micropub.biology.000420
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.