
- Home
- Protocols

-

Preparation of libraries for yeast ribosome footprints
Last updated date: Jul 2, 2021 Views: 963 Forks: 0
Methods
Yeast cell harvesting by filtration and flash freeze
- Set up filtration apparatus with a Whatman cellulose nitrate 0.45 µm filter membrane and connect to a direct vacuum pump.
- Place a tube rack in an ice bucket and fill it with liquid nitrogen. Puncture holes into the bottom of a 50 mL conical tube using an 18-G needle and place it on the tube rack. The level of liquid nitrogen needs to be at least 3-4 inches deep to submerge half of a 50mL conical tube.
- Pour ~800 mL of the yeast culture into filtration apparatus. While the media is filtering, scrape with a plastic spatula along the surface of membrane till a clump of yeast cells accumulate on the spatula (see Note 1).
- Plunge the spatula into the tube submerged in liquid nitrogen and flick off the cells off with a pre-chilled pipette tip (see Note 2).
- Add 1 mL of footprint lysis buffer A into the tube full of liquid nitrogen.
- Cap the conical tube and store at -80˚C. Liquid nitrogen will drain through the holes on the bottom of the tube.
Lyse by freezer mill and polysome isolation
- Set up a freezer mill. Prechill a sample prep grinder small vial on dry ice.
- Place frozen yeast sample, lysis buffer, and a grinding rod inside vial.
- Run freezer mill with settings 1 min on and 1 min off for 8 cycles.
- Take vial out of freezer mill. Remove cap. Immediately pour frozen ground powder into 22 mL of pre-chilled footprint lysis buffer A in a labeled 50 mL conical tube (see Note 3).
- Vortex to thaw frozen lysate powder in the conical tube.
- Centrifuge at 4˚C (15,000 rpm, 15 min). Transfer supernatant into a fresh 50 mL conical tube with a serological pipette.
- Layer supernatant from Step 12 on the top of 3 mL sucrose cushion solution in a polycarbonate bottle (see Note 4).
- Centrifuge in a Type 70 Ti rotor at 4˚C (60,000 rpm, 106 min).
- Remove supernatant from ribosome pellets with a 10 mL serological pipette (see Note 5).
- Wash ribosome pellets once gently with 0.5 mL cold footprint lysis bufferB.
- Resuspend ribosome pellets in 1 mL cold footprint lysis buffer B with gentle pipetting on ice.
- Determine RNA concentration using absorption.
- Resuspended ribosome pellets can be flash frozen and kept at -80˚C.
Monosome isolation by sucrose gradient
- Digest 350 µg of pelleted polysomes from Step 19 with 500 U of Ambion TM RNase I in 300 µL footprint lysis buffer B on a thermomixer at 25 ˚C (500 rpm, 1 hr).
- Stop RNase I reaction by adding 6,000 U of SUPERase*In and place digested samples on ice.
- During RNase I digestion, use a gradient maker to prepare 15- 50% sucrose gradient solution in SW41 polyclear tubes.
- Layer digested solution from Step 21 onto 15-50% sucrose gradient solution.
- Centrifuge in a SW41 rotor at 4˚C (40,000 rpm, 2 hrs).
- Use a gradient fractionator to isolate monosome fractions. Monosome fractions can be stored at -80˚C for 6 months.
- Preheat 0.75 mL of acid phenol in 2-mL screwcap tubes at 65 ˚C.
- Add 57 µL of 20% SDS solution per mL of monosome fractions from Step 25 and heat to 65 ˚C on a thermomixer to dissolve SDS.
Add heated monosome fractions to preheated aid phenol and incubate at 65˚C on a thermomixer shaking at 1,300 rpm for 5 min (see Note 6). - Chill samples on ice for 3 min. Centrifuge at room temperature (15,000 rpm, 3 min).
- Pull aqueous (bottom) layer and add to a 2-mL screwcap tube containing 0.7 mL acid phenol at room temperature.
- Vortex samples for 3 min at room temperature. Centrifuge at room temperature (15,000 rpm, 3 min).
- Pull aqueous (top) layer and add to a 2-mL screwcap tube containing 0.6 mLchloroform.
- Vortex samples for 3 min at room temperature. Centrifuge at room temperature (15,000 rpm, 3 min).
- Pull aqueous (top) layer. Add 3 M NaOAc (pH 5.5) to 0.3 M. Then add equal volume of isopropanol and 2 µL of GlycoBlue.
- Precipitate RNA samples on dry ice for 30 min. Centrifuge at 4 ˚C (15,000 rpm, 30 min).
- Discard supernatant and wash blue RNA pellets with 500 µL of 80 % ice-cold ethanol, and air-dry for 15 min.
- Resuspend RNA pellets in 10 -20 µL of 10 mM Tis-Cl (pH 8). Quantify RNA using absorption.
Construction of a sequencing library from ribosome-protected mRNA fragments Size select ribosome-protected mRNA fragments
- Mix 4 µg of RNA from Step 37 with equal volume of RNA loading buffer.
- Mix 2 µL of size control ladders (15 nt and 34 nt RNA) with equal volume of RNA loading buffer.
- Heat samples to 80˚C for 2 min and then immediately place on ice.
- Load samples on a 15% TBE-urea polyacrylamide gel and run at 340 V for 20 min.
- Puncture holes into the bottom of a 0.5 mL PCR tube with an 18-G needle. Also prepare a punctured 0.5 mL PCR tube for control ladders.
- Stain gel with 50 mL gel staining solution for 5 min with shaking at room temperature.
Image gel with a Typhoon biomolecular imager and excise footprints between 15 and 34 nt as shown in Figure 2. Place gel slices into a punctured 0.5 mL PCR tube from Step 42. Also excise control ladders to keep as a size control (see Note 7). - Place PCR tubes full of gel slices from Step 43 into 1.5 mL RNase-free eppendorfs and centrifuge at room temperature (15,000 rpm, 3 min) to shred the gel slices into the eppendorfs.
- Add 450 µL RNA elution buffer to gel slices and elute on a nutator at 4˚C overnight.
- Transfer gel mixture to a Costar Spin-X column. Centrifuge at 4˚C (15,000 rpm, 3 min).
- Transfer eluates from Step 46 into a new RNase-free Eppendorf. Add 2 µL GlycoBlue and 450 µL isopropanol. Mix well and chill on dry ice for 30 min.
- Centrifuge at 4˚C (15,000 rpm, 30 min) to pellet isolated RPFs.
- Remove supernatant, wash pellets with 500 µL of 80 % ice-cold ethanol, and air-dry for 15 min.
- Resuspend pellets in 3.5 µL of 10 mM Tris-Cl (pH 8) and transfer RNA and size control samples to 0.2-mL thin-walled PCR tubes
Dephosphorylation
- Heat RNA and size control samples to 80 ˚C for 2 min and place on ice.
- Add 1 µL of Dephosphorylation reaction mix and 0.5 µL of T4 polynucleotide kinase.
- Incubate at 37 ˚C for 1 hr in a thermocycler.
Linker ligation
- Add 4.5 µL of Ligation reaction mix directly to dephosphorylated sample from Step 52 (see Note 8).
- Add 0.5 µL of T4 RNA ligase and incubate at 37 ˚C for 3 hrs in a thermocycler.
- Clean up each reaction with a Zymo oligo clean and concentrate kit (see Note 9).
Ribosomal RNA depletion
- Deplete ribosomal rRNA from samples using a RiboPool yeast rRNA depletion kit according to the manufacturer’s instructions. (see Notes 10 and 11)
- Clean up each reaction with a Zymo oligo clean and concentration kit and elute RNA in 15 µL of nuclease-free water.
Reverse transcription
- Mix 10 µL of RNA and size control samples from Step 58 with 1.25 µL of RT primer, heat up to 80˚C for 2 min, and place on ice.
- Add 7.75 µL of RT reaction mix and then 1 µL of Superscript III.
- Incubate samples at 55˚C for 30 min in a thermocycler.
- Add 2 µL of 1 N NaOH to each sample and incubate at 95 ˚C for 15 min in a thermocycler.
- Clean up RNA and size control samples with a Zymo oligo clean and concentration kit and elute in 10 µL of nuclease-free water.
- Mix the eluate with equal volume of RNA loading buffer.
- Heat samples to 80˚C for 2 min and then immediately place on ice.
- Load samples on a 10% TBE-urea polyacrylamide gel and run at 340 V for 1 hr.
- Excise reactions products from gels as described in Steps 43- 45. Use size control samples as a guide to excise reverse transcription products as shown in Figure 3.
- Shred gel slices as described in Step 44.
- Add 450 µL DNA elution buffer to shredded gel slices and elute on a nutator at room temperature overnight.
- Transfer gel mixture to a Costar Spin-X column. Centrifuge at room temperature (15,000 rpm, 3 min).
- Precipitate reaction products as described in Steps 47-49.
Circularization
- Resuspend pellets in 15 µL of Circularization reaction mix and incubate at 60˚C for 2 hrs on a thermomixer.
- Heat up samples to 80˚C for 10 min to inactivate the enzyme. Circularized DNA can be stored at -80˚C for 3 months.
Preparative PCR
1. Add 2 µL of circularized DNA sample and 2 µL barcoded PCR primer to 36 µL of PCR master mix.
2. Aliquot 20 µL sample master mix into two 0.2-mL thin-walled PCR tubes. Each tube will undergo different numbers of PCR cycles.
3. Perform PCR as follows (see Note 12):
| 1 cycle: | 98˚C | 30 sec |
| N cycles: | 98˚C | 10 sec |
| 65˚C | 10 sec | |
| 72˚C | 5 sec | |
| 1 cycle: | 16˚C | hold |
4. Add 4 µL of DNA loading buffer to eachsample.
5. Run an 8% TBE polyacrylamide gel at 340 V for 1 hr (Figure 4).
6. Stain gel with gel staining solution for 5 min with shaking at room temperature. Excise PCR products between 160 and 190 bp. 7. Shred gel slices and precipitate PCR products as described in Steps 69-71.
8. Resuspend PCR products in 6 µL of 10 mM Tris-Cl (pH 8). Dilute PCR products if necessary.
9. Quantify DNA concentration on a Bioanalyzer with a DNA high-sensitivity chip according to manufacturer’s instructions. The 10. size range of PCR products should be around 160- 190 bp.
11. Submit quantified PCR products for Illumina sequencing. We recommend 50 bp sequencing on a NextSeq 2000 or NovaSeq 6000 machine.
References
- Dever, T. E., Dinman, J. D. & Green, R. Translation Elongation and Recoding in Eukaryotes. Cold Spring Harb Perspect Biol (2018) doi:10.1101/cshperspect.a032649.
- Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. & Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science (80-. ). 324, 218–223 (2009).
- Ingolia, N. T., Lareau, L. F. & Weissman, J. S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789– 802 (2011).
- Wolin, S. L. & Walter, P. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J. 7, 3559–69 (1988).
- Lareau, L. F., Hite, D. H., Hogan, G. J. & Brown, P. O. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. Elife 3, e01257 (2014).
- Wu, C. C.-C., Zinshteyn, B., Wehner, K. A. & Green, R. High-Resolution Ribosome Profiling Defines Discrete Ribosome Elongation States and Translational Regulation during Cellular Stress. Mol. Cell 73, 959-970.e5 (2019).
- Budkevich, T. et al. Structure and dynamics of the mammalian ribosomal pretranslocation complex. Mol Cell 44, 214–224 (2011).
- Jenner, L. et al. Structural basis for potent inhibitory activity of the antibiotic tigecycline during protein synthesis. Proc Natl Acad Sci U S A 110, 3812–3816 (2013).
- Jiang, H., Lei, R., Ding, S. W. & Zhu, S. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics 15, 182 (2014).
- Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
- Schuller, A. P., Wu, C. C., Dever, T. E., Buskirk, A. R. & Green, R. eIF5A Functions Globally in Translation Elongation and Termination. Mol. Cell 66, (2017).
- dos Reis, M., Savva, R. & Wernisch, L. Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res 32, 5036–5044 (2004).
- McGlincy, N. J. & Ingolia, N. T. Transcriptome-wide measurement of translation by ribosome profiling. Methods 126, 112–129 (2017).
- Pfeffer, S., Lagos-Quintana, M. & Tuschl, T. Cloning of small RNA molecules. Curr. Protoc. Mol. Biol. (2005) doi:10.1002/0471142727.mb2604s72.
- Zinshteyn, B., Wangen, J. R., Boyang, H. U. A. & Green, R. Nuclease-mediated depletion biases in ribosome footprint profiling libraries. RNA (2020) doi:10.1261/rna.075523.120.
- Guydosh, N. R. & Green, R. Dom34 Rescues Ribosomes in 3ʹ Untranslated Regions. Cell 156, 950–962 (2014).
- Gu, W. et al. Depletion of Abundant Sequences by Hybridization (DASH): Using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol. (2016) doi:10.1186/s13059-016-0904-5.
- Han, P. et al. Genome-wide Survey of Ribosome Collision. Cell Rep. 31, 107610 (2020).
Figure Legends
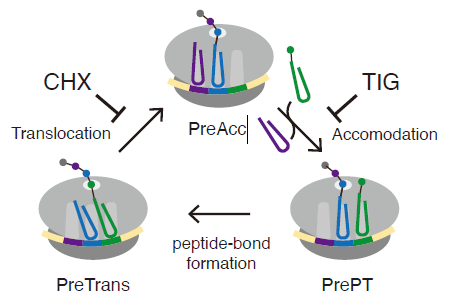
Figure 1. Eukaryotic elongation cycle
A schematic representation of the eukaryotic elongation cycle. Ribosome functional states are abbreviated: PreAcc, pre-accommodation; PrePT, pre-peptide bond formation; PreTrans, pre- translocation. Cycloheximide (CHX) and tigecycline (TIG) are used to inhibit translocation and accommodation, respectively.

Figure 2. Ribosome footprint size-selection gel
An image of size selection on a 15% TBE-urea gel from RPFs prepared from S. cerevisiae BY4171. Size control ladders (15 nt and 34 nt) are indicated. Areas marked with dashed lines indicate regions to be excised.
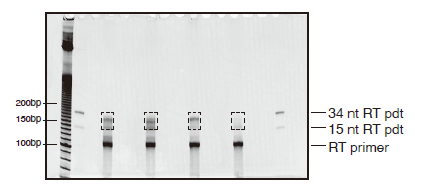
Figure 3. Reverse transcription gel
A representative gel image of reverse transcription reactions on a 10% TBE-urea gel. 10-bp DNA ladders, 15 nt-RT and 34 nt-RT products are used to guide gel excision. Dashed boxes indicate areas to be excised. Unreacted RT primer is also indicated and should be avoided during gel excision.

Figure 4. Preparative PCR gel
A representative gel image of preparative PCR reactions on an 8% polyacrylamide native gel. 10bp DNA ladders are included to guide gel excision. Dashed boxes indicate gel areas to be excised. Asterisk marks high-molecular weight PCR products. We do not recommend excising PCR products from reactions that have high-molecular weight products.
- Green, R and Wu, C(2021). Preparation of libraries for yeast ribosome footprints. Bio-protocol Preprint. bio-protocol.org/prep1244.
- D'Orazio, K. N., Wu, C. C., Sinha, N., Loll-Krippleber, R., Brown, G. W. and Green, R.(2019). The endonuclease Cue2 cleaves mRNAs at stalled ribosomes during No Go Decay. eLife. DOI: 10.7554/eLife.49117
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.