
- Home
- Protocols

-

Visualizing, Binning, and Refining of Metagenome-assembled Genomes (MAGs) with Anvi’o
Published: May 20, 2023 DOI: 10.21769/BioProtoc.4684 Views: 683
Reviewed by: Varun KesherwaniHassan Rasouli
Abstract
High throughput ‘omics technologies generate huge datasets that need to be properly analyzed in order to decipher the biological implications. The workflow of handling such datasets must be user friendly to facilitate rapid analysis. Here, we demonstrate the use of the Anvi’o workflow, which is a visualization platform that allows for advanced analysis of metagenomics data. In this protocol, we provide the pre-packaged plant-microbiome dataset. Then, we use the dataset to visualize and perform manual binning and refinement of metagenome-assembled genomes (MAGs). Anvi’o works with an easy-to-use interface and also helps users to test and implement research ideas in a timely manner.
Keywords: MAGsBackground
Shotgun metagenomics is a popular approach for studying microbial community, diversity, and functional potential (Handelsman et al., 1998; Sogin et al., 2006). These types of high throughput sequencing technologies generate a huge amount of metagenomic data that need to be analyzed properly to understand the biological implications (D'Argenio, 2018). Assembling short reads into contigs is essential to improve annotations, and the compatible genomic binning technique can further link unconnected contigs into biologically meaningful units (Tyson et al., 2004; Venter et al., 2004). Metagenomic binning is the process by which metagenomic sequences can be grouped using the organisms of origin. This leads to reconstruction of genomes, which can be used in downstream analyses (Turaev and Rattei, 2016; Wang and Jia, 2016; Quince et al., 2017; Nissen et al., 2021). With existing automated software pipelines, studies have successfully implemented assembly and binning approaches to draft genomes that are near complete in nature (Hess et al., 2011; Alneberg et al., 2014; Wu et al., 2014; Kang et al., 2015; Raveh-Sadka et al., 2015). However, the existing pipelines do not provide the contigs distribution across samples (Sharon et al., 2013; Alneberg et al., 2014). At this juncture, there is a need for an easy visualization interface–based workflow that is competent for analyzing such large metagenomic datasets. Anvi’o (advanced analysis and visualization platform for ‘omics data) provides an easy management of contigs, where both manual or automatic genome bins identifications and curations are possible. This pipeline is also capable of generating a unified display of inferred taxonomy and GC-content with the contig numbers in different samples (Eren et al., 2015).
Software
Anvi’o v7 (https://merenlab.org/software/anvio/)
Installing Anvi’o:
Conda setup: If the conda is not installed in the system, it is necessary to open a terminal such as iTerm.
Command:
conda install
To verify whether you already have conda installed, copy and paste the following command into your terminal:
Command:
conda --version
Always make sure that you work in an up-to-date conda environment by using the following command:
Command:
conda update conda
Anvi’o environment setup
Create a new conda environment using the command:
Command:
conda create -y --name anvio-7.1 python=3.6
Then, activate it using the command:
Command:
conda activate anvio-7.1
Installing Anvi’o
The first step is to download the python source package for the Anvi’o release using the following command:
Command:
curl -L https://github.com/merenlab/anvio/releases/download/v7.1/anvio-7.1.tar.gz \
--output anvio-7.1.tar.gz
Then, use the following command to install Anvi’o:
Command:
pip install anvio-7.1.tar.gz
The users should note that the installation of Anvi’o is user friendly but may take a long time to finish and is computationally intensive.
Updating Anvi’o databases
If the Anvi’o databases are not compatible with the latest versions of Anvi’o, there are options to update the Anvi’o databases.
The safest option is to use:
Command:
anvi-migrate --migrate-dbs-safely *.db
When using this option, Anvi’o will generate a backup of a copy of each database. If there is an error during the migration, the Anvi’o will let the users know about what went wrong and will be able to restore the original database from the copy it made.
Another option is to use:
Command:
anvi-migrate --migrate-dbs-quickly *.db
This option does not create any backup files but might be useful when there are a lot of databases to migrate.
Data availability
The data can be accessed at https://figshare.com/s/acc45cb6fb5cbd819d69 and https://github.com/Bio-protocol/bioprotocol_2104072.git
Case study
Downloading the pre-packaged plant-microbiome dataset
The plant-microbiome data pack can be downloaded from https://figshare.com/s/acc45cb6fb5cbd819d69.
Following are some details about the plant-microbiome data pack:
In the dataset directory, you will see that the data pack contains an Anvi'o merged profile database (that describes six metagenomes: three from plants and three from fecal samples), an Anvi'o contigs database, and additional extra data that are required by various sections in this tutorial. Here are some basic descriptions of several of these files, as well as how they were created:
The profile and contigs databases. We produced the Anvi'o contigs database utilizing the program anvi-gen-contigs-database. This Anvi'o database keeps all the information that is associated with the contigs: k-mer frequencies for each contig, open reading frames positions, taxonomic and functional annotation of genes, among others. We also used the tool anvi-profile to create a merged Anvi'o profile database. Anvi'o profile databases store sample-specific information on contigs. Each profile database is linked to a contigs database, and Anvi'o uses the program anvi-merge to merge single profiles that are linked to the same contigs database into an Anvi'o merged profile.
Single-copy core genes in contigs. Among the contigs, we utilized the program anvi-run-hmms to find single-copy core genes for Bacteria, Archaea, and Eukarya, as well as ribosomal RNA sequences. The contigs database stores all these results as well. This data enables us to learn the completeness and redundancy estimations of freshly detected bins using the interactive interface. Note that if all single-copy core genes for a given domain are discovered once in the chosen bin, the completion rate is 100% and the redundancy rate is 0%. The redundancy score will rise if a few genes are discovered several times. In case a few genes are missing, the completion value will be reduced.
Assigning functions towards genes. We performed anvi-run-ncbi-cogs and anvi-run-kegg-kofams and stored gene functions results in the contigs database.
Genome-resolved metagenomics
This tutorial uses a plant-microbiome dataset to discuss genome-resolved metagenomics (with a focus on manual binning). You will be able to do the following by the end of this tutorial:
Learn how to use the interactive binning interface.
Examine contigs in relation to their metagenomic signal.
Perform manual binning to characterize bins.
Summarize the findings of manual binning for use in subsequent studies.
Curate bins manually for the purpose of quality control.
FASTA and BAM files of the contigs are used in a typical Anvi'o genome-resolved metagenomic approach. In this tutorial, we will start at the stage in the workflow where you have generated Anvi'o contigs and profile databases using your FASTA and BAM files (Figure 1).
Figure 1. Workflow for the users to follow to obtain the results for this protocolLet us look at the merged profile database for the plant-microbiome dataset metagenome using the files in the data pack directory.
The following anti-interactive command on the merged profile database will initiate the Anvi’o interactive interface:
Command:
anvi-interactive -p PROFILE.db -c CONTIGS.db -C concoct
After you click “draw,” the Anvi’o interactive interface should greet you with the following display (Figure 2):
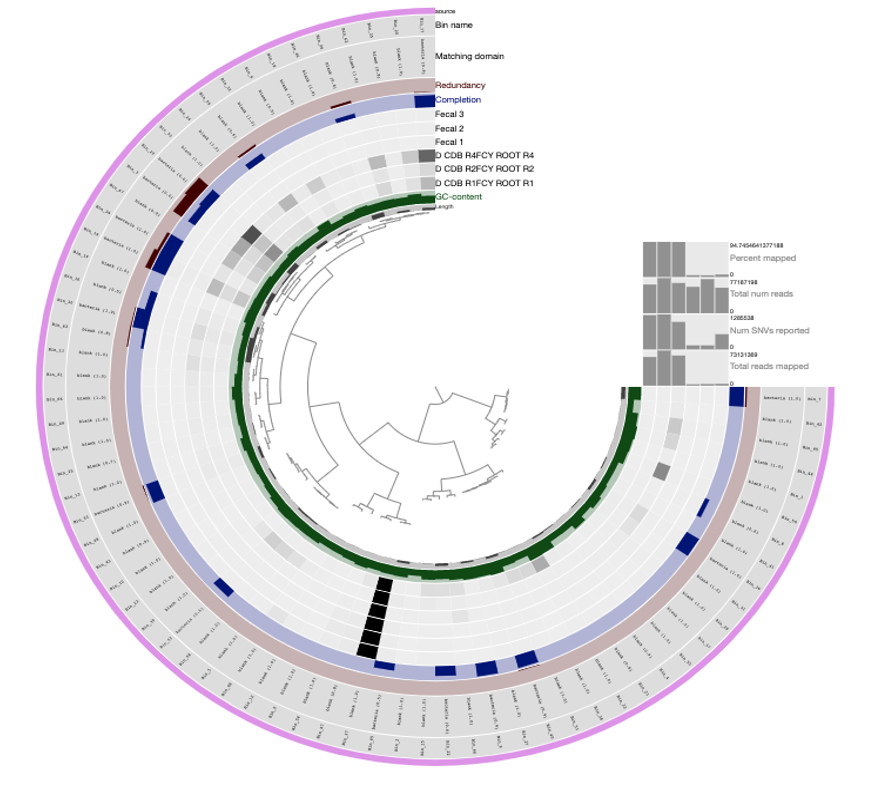
Figure 2. The Anvi’o interactive interface, showing the merged profile of three plant and fecal metagenomesClose the window, return to the terminal, and hit CTRL + C to stop the server.
Taking a look at the binning findings
We are interested in putting our metagenomes into context with the genomes we have retrieved through binning. Comprehending the quantitative distribution patterns of the genomes in a collection, obtaining a table of function discovered, or summarizing our bins as separate FASTA files are all crucial for the downstream analysis of any binning workflow. We used anvi-summarize to summarize any collection saved in a Anvi'o profile database. You will have a static HTML page that you can visualize on any computer (Figure 3).
Command:
anvi-summarize -p PROFILE.db \
-c CONTIGS.db \
-C concoct \
-o SUMMARY
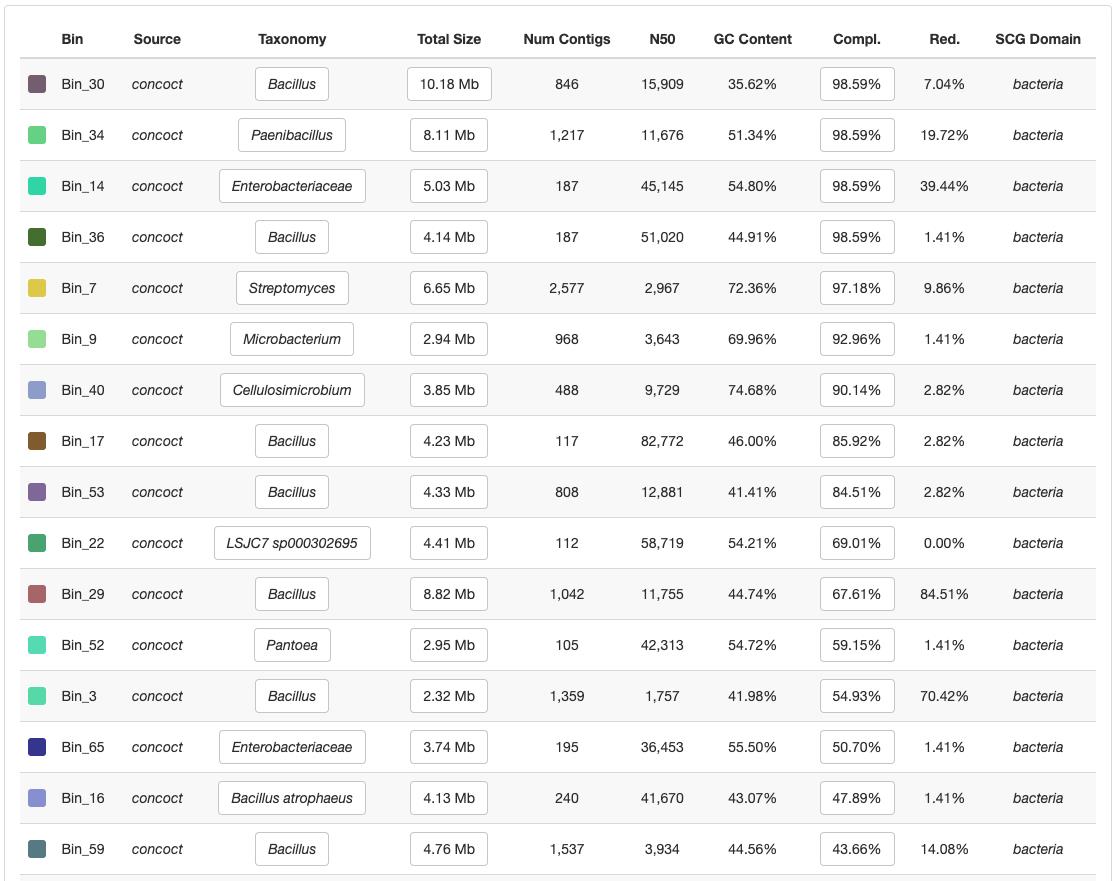
Figure 3. Summary of binsManual curation to refine individual MAGs
We can now go through a round of manual binning. A couple of binning pointers:
It is not necessary to bin all contigs. Instead, look for bins that correlate to a real genome with high completion values.
Avoid bins with redundancy higher than 10%. Those are most likely contaminated.
In this profile, we identified 69 bins after the auto-binning protocol. In Anvi’o, a collection describes one or more bins. Each bin describes single or multiple contigs. Please keep all the bins together as a collection. You can name your collection whatever you like. Individual bins can be visualized and refined if necessary to improve the quality of the MAGs collection. We used the program anvi-refine for this.
Command:
anvi-refine -p PROFILE.db \
-c CONTIGS.db \
-C concoct \
-b Bin_34
Contigs from a single bin are now displayed in the interactive interface (Figure 4A). During the curation step, several clustering algorithms can be used to detect outliers and determine if they are contaminants. We can select and remove the contigs that we do not want to keep in the bin, and then we can store the updated set of contigs in the database using the “bins” panel. In this example of MAG (Bin_34) we curated, we removed four contigs (Figure 4B):
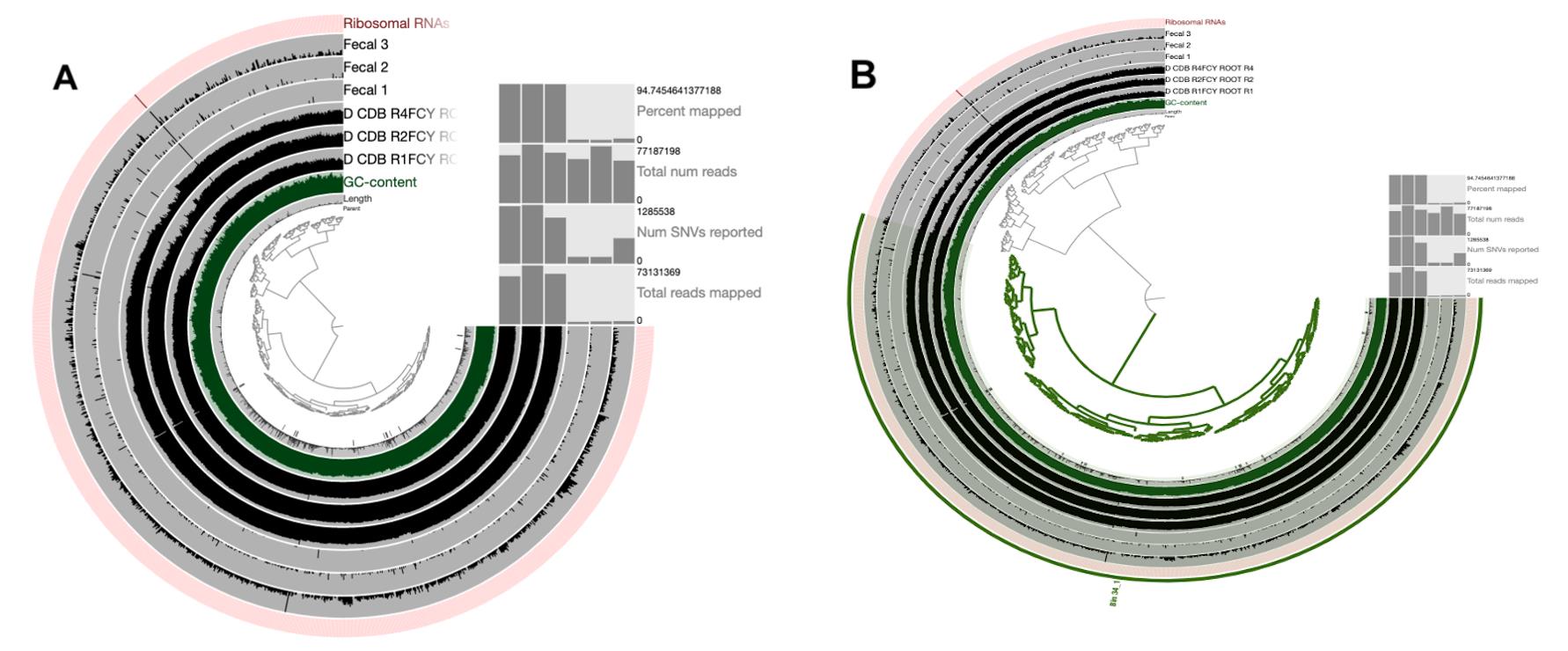
Figure 4. Interactive interface displaying contigs from a single bin. A. Display of contigs from a single bin before manual curation. B. Curation of Bin_34, removing four contigs to achieve a higher level of completion and redundancy. Henceforth, we will rename Bin_34 as a metagenome-assembled genome (MAG).Figure 4B can be obtained by clicking the branches to add into separate groups. After refining, the contamination reduces to much lower levels. The users can also refer to the video provided with the protocol to follow the steps required to curate the bin. The collection will be changed by storing the refined new bin in the database. This is a straightforward example. However, improving a given MAG can take hours in some circumstances.
Renaming bins in your collection
From the summary file, you can see that bin names are currently arbitrary, and we frequently find it helpful to impose some order at this step. This is a particularly beneficial method when the goal is to eventually merge numerous binning efforts. We use the tool anvi-rename-bins to rename bins:
Command:
anvi-rename-bins -p PROFILE.db \
-c CONTIGS.db \
--collection-to-read concoct \
--collection-to-write MAGs \
--call-MAGs \
--prefix Soil \
--report-file rename-bins-report.txt
With those parameters, a new MAG collection will be formed, in which (1) bins with a completion >70% are designated as MAGs (metagenome-assembled genomes), and (2) bins and MAGs are given a prefix and renamed based on the difference between redundancy and completion. The users can customize the parameters involving the completion and redundancy. We recommend bins with completions >70% and redundancy <10%.
At this point we can summarize the new collection using the program anvi-summarize
Command:
anvi-summarize -p PROFILE.db \
-c CONTIGS.db \
-C MAGs \
-o SUMMARY_AFTER_RENAME
Now, double-click on the file index.html to visualize the outputs that are currently in the newly created folder SUMMARY_MAGs (Figure 5).
We get the following MAGs:
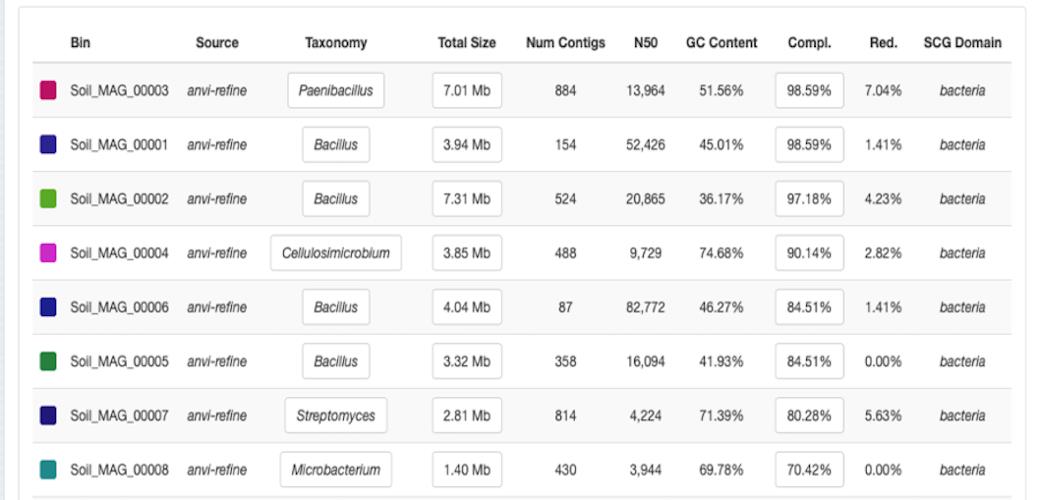
Figure 5. Summary of the new collection of MAGs. The takeaway point here is that it is possible to enhance the results through manual refining when automatic binning techniques may produce poorly identified bins.
Result interpretation
Proper manual refinement of MAGs is necessary to elucidate meaningful biological implications. In this protocol, we used a plant-microbiome dataset to identify 69 bins and then curated the bins manually to remove the outliers and contaminants. It is recommended to perform manual refining after automatic binning to recover higher quality MAGs.
Discussion
Anvi’o is an easy-to-use interface that enables the user to visualize, perform binning, and refine MAGs. This protocol enables users to carry out the entire workflow and provides a scope to improvise the method for other datasets.
Acknowledgments
Soumyadev Sarkar acknowledges the National Science Foundation EPSCoR for his research grant. This protocol is based on using Anvi’o (Eren et al., 2015 and 2021). This material is based upon work supported by the National Science Foundation under Award No. OIA‐1656006 and matching support from the State of Kansas through the Kansas Board of Regents.
Competing interests
The authors declare no competing interest.
References
- Alneberg, J., Bjarnason, B. S., de Bruijn, I., Schirmer, M., Quick, J., Ijaz, U. Z., Lahti, L., Loman, N. J., Andersson, A. F. and Quince, C. (2014). Binning metagenomic contigs by coverage and composition. Nat Methods 11(11): 1144-1146.
- D'Argenio, V. (2018). The High-Throughput Analyses Era: Are We Ready for the Data Struggle? High Throughput 7(1).
- Eren, A. M., Esen, O. C., Quince, C., Vineis, J. H., Morrison, H. G., Sogin, M. L. and Delmont, T. O. (2015). Anvi'o: an advanced analysis and visualization platform for 'omics data. PeerJ 3: e1319.
- Eren, A. M., Kiefl, E., Shaiber, A., Veseli, I., Miller, S. E., Schechter, M. S., Fink, I., Pan, J. N., Yousef, M., Fogarty, E. C., et al. (2021). Community-led, integrated, reproducible multi-omics with anvi'o. Nat Microbiol 6(1): 3-6.
- Handelsman, J., Rondon, M. R., Brady, S. F., Clardy, J. and Goodman, R. M. (1998). Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products. Chem Biol 5(10): R245-249.
- Hess, M., Sczyrba, A., Egan, R., Kim, T. W., Chokhawala, H., Schroth, G., Luo, S., Clark, D. S., Chen, F., Zhang, T., et al. (2011). Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 331(6016): 463-467.
- Kang, D. D., Froula, J., Egan, R. and Wang, Z. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3: e1165.
- Nissen, J. N., Johansen, J., Allesoe, R. L., Sonderby, C. K., Armenteros, J. J. A., Gronbech, C. H., Jensen, L. J., Nielsen, H. B., Petersen, T. N., Winther, O. et al. (2021). Improved metagenome binning and assembly using deep variational autoencoders.Nat Biotechnol 39(5): 555-560.
- Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J. and Segata, N. (2017). Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 35(9): 833-844.
- Raveh-Sadka, T., Thomas, B. C., Singh, A., Firek, B., Brooks, B., Castelle, C. J., Sharon, I., Baker, R., Good, M., Morowitz, M. J. et al. (2015). Gut bacteria are rarely shared by co-hospitalized premature infants, regardless of necrotizing enterocolitis development. Elife 4: e05477.
- Sogin, M. L., Morrison, H. G., Huber, J. A., Mark Welch, D., Huse, S. M., Neal, P. R., Arrieta, J. M. and Herndl, G. J. (2006). Microbial diversity in the deep sea and the underexplored "rare biosphere". Proc Natl Acad Sci U S A 103(32): 12115-12120.
- Sharon, I., Morowitz, M. J., Thomas, B. C., Costello, E. K., Relman, D. A. and Banfield, J. F. (2013). Time series community genomics analysis reveals rapid shifts in bacterial species, strains, and phage during infant gut colonization. Genome Res 23(1): 111-120.
- Turaev, D. and Rattei, T. (2016). High definition for systems biology of microbial communities: metagenomics gets genome-centric and strain-resolved.Curr Opin Biotechnol 39: 174-181.
- Tyson, G. W., Chapman, J., Hugenholtz, P., Allen, E. E., Ram, R. J., Richardson, P. M., Solovyev, V. V., Rubin, E. M., Rokhsar, D. S. and Banfield, J. F. (2004). Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 428(6978): 37-43.
- Venter, J. C., Remington, K., Heidelberg, J. F., Halpern, A. L., Rusch, D., Eisen, J. A., Wu, D., Paulsen, I., Nelson, K. E., Nelson, W., et al. (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science 304(5667): 66-74.
- Wang, J., and Jia, H. (2016). Metagenome-Wide Association Studies: Fine-Mining the Microbiome. Nat Rev Microbiol 14(8): 508-522.
- Wu, Y. W., Tang, Y. H., Tringe, S. G., Simmons, B. A. and Singer, S. W. (2014). MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2: 26.
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
Category
Plant Science > Plant molecular biology > DNA > DNA sequencing
Systems Biology > Genomics > Functional genomics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.