
- Home
- Protocols

-

The Canu Genome Assembly Pipeline Using Nanopore Long Reads
Published: Sep 5, 2022 DOI: 10.21769/BioProtoc.4501 Views: 1561
Reviewed by: Thibaud T. RenaultHassan RasouliHainan Zhao
Abstract
Long sequencing reads have greatly improved assemblies of genomes with all sizes. The current Oxford Nanopore technology can regularly produce reads longer than 20 kb with less than 10% sequencing errors. To use long reads that contain relatively high errors, algorithms have been developed for genome assembly and sequence polishing. This pipeline shows the process of the genome assembly from raw data to polished assembled contigs, including basecalling from Nanopore raw reads using the Guppy basecaller, genome assembly with the Canu genome assembler, and sequence polishing using both Nanopore long reads and Illumina short reads, with Nanopolish and Pilon, respectively. Small fungal datasets were used to illustrate the pipeline. The pipeline has been demonstrated to produce high-quality genome assemblies of fungal and plant genomes.
Graphical abstract:
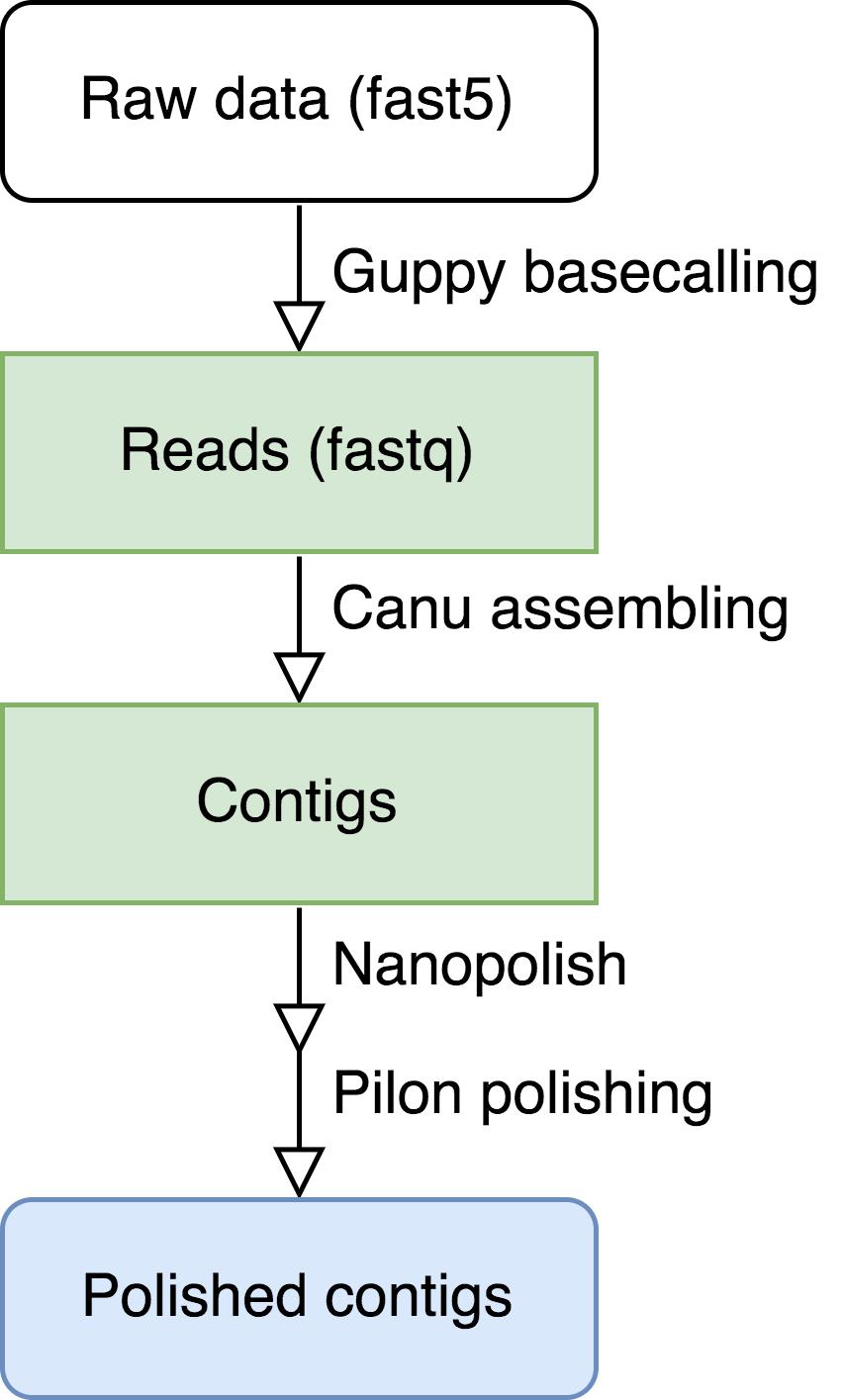
Flowchart from raw data to genome assembly.
The pipeline includes three major steps: basecalling, read assembly, and polishing of assembled contigs. Software Guppy, Canu, Nanopolish, and Pilon are used with other bioinformatics tools in this pipeline.
Background
The genome assembly has been dramatically improved with newly developed long-read sequencing technologies. Oxford Nanopore sequencing has been used for genome assemblies of a wide range of species, including the large and complex maize genome (Lin et al., 2021). Using Nanopore long-read sequencing technology, single-stranded DNA or RNA molecules pass through a nanopore protein affecting the ionic current during the movement (Jain et al., 2016). The electrical signals captured by the sensors are converted to sequencing reads with a basecaller (Jain et al., 2016; Wick et al., 2019). Output sequencing reads are typically within a range of lengths, largely depending on the size of input molecular fragments. Nanopore long reads have a relatively high rate of errors, dominated by small insertions or deletions. Recently, the error rate has been markedly reduced with optimized library chemistry and improved basecalling algorithms (Wang et al., 2021). Multiple genome assemblers have been developed to take advantage of long reads, considering high error rates (Li, 2016; Koren et al., 2017; Kolmogorov et al., 2019). Canu has been demonstrated to be a reliable assembler that corrects/trims reads and assembles cleaner reads. Here, we employed Canu for genome assembly using Oxford Nanopore long reads, followed by contig polishing with Nanopolish and Pilon (Walker et al., 2014; Loman et al., 2015). We demonstrate the procedure using read data from a chromosomal segment of a fungal genome (Figure 1) (Peng et al., 2019; He et al., 2020). The codes and parameters used here have been shown to be applicable for assemblies of plant genomes (Lin et al., 2021).
Software and Data sets
Software
Guppy Basecaller [(Oxford Nanopore Technologies, 2022); version 6.0.6; https://community.nanoporetech.com]
Canu [(Koren et al., 2017); version 2.2; https://github.com/marbl/canu]
Nanopolish [(Loman et al., 2015); version 0.13.3; https://github.com/jts/nanopolish]
Pilon [(Walker et al., 2014); version 1.24; https://github.com/broadinstitute/pilon]
Minimap2 [(Li, 2018b); version 2.17; https://github.com/lh3/minimap2]
BWA [(Li, 2013); version 0.7.17; https://github.com/lh3/bwa]
SAMtools [(Li et al., 2009); version 1.12; https://github.com/samtools/samtools]
MUMmer4 [(Marçais et al., 2018); version 4.0.0; https://mummer4.github.io/]
Seqtk [(Li, 2018a); version 1.3; https://github.com/lh3/seqtk]
Installation
Download the bio-protocol repository from GitHub.
git clone https://github.com/Bio-protocol/Nanopore_genome_assembly.git
cd Nanopore_genome_assembly
Download Guppy from the Oxford Nanopore Community webpage. Due to the copyright, the website for downloading the software is not provided. Users can register at nanoporetech.com to access the latest version of Guppy software. Here, we show the example for the installation of Guppy version 6.0.6.# Example for downloading Guppy (version 6.0.6)
pushd lib
curl -L -o ont-guppy-cpu_6.0.6_linux64.tar.gz <Guppy downloading site>tar -xvzf ont-guppy-cpu_6.0.6_linux64.tar.gz
rm ont-guppy-cpu_6.0.6_linux64.tar.gz
popdDownload Pilon.
For using Pilon Java script, no installation is required.
pushd lib
pilon_site=https://github.com/broadinstitute/pilon/releases/download/v1.24/pilon-1.24.jarcurl -L -o pilon-1.24.jar $pilon_site
popd
Most tools can be installed through conda (https://docs.conda.io). Create a conda environment and install packages.
conda create -n npasm
conda activate npasm
conda install -c bioconda -c conda-forge -c defaults canu=2.2 nanopolish=0.13.2
conda install -c bioconda samtools seqtk minimap2 bwa mummer4
conda deactivate
Input data
Fast5 is a binary HDF5 file format for storing the raw electrical signal level data of Oxford Nanopore sequencing reads (Wang et al., 2021). To produce a small dataset, a subset of fast5 data was extracted from the Nanopore Whole Genome Sequencing (WGS) data of the wheat blast fungal isolate B71, using the fast5_subset tool (nanoporetech). In detail, B71 Nanopore fastq reads (SRR12459118) called from fast5 were first aligned to the reference genome (B71Ref1.5) using minimap2. We then extracted fast5 data of half of reads mapped to the region from 1,000,000 to 1,050,000 on chromosome 5 (chr5:1000000-1050000) of B71Ref1.5 using fast5_subset, which were saved in the following three fast5 files. These fast5 files are used for Guppy basecalling and Nanopolish polishing.
| input/ont_fast5/fast5/B71example0.fast5 input/ont_fast5/fast5/B71example1.fast5 input/ont_fast5/fast5/B71example2.fast5 |
Illumina read data mapped to the same chromosome 5 region were extracted from the trimmed paired-end 250 bp Illumina reads of B71 (SRR6232156) using seqtk. Briefly, Illumina WGS reads were mapped to B71Ref1.5 using BWA-MEM. Reads mapped to the region chr5:1000000–1050000 of B71Ref1.5 were extracted. The extracted paired-end reads are used for Pilon polishing.
| input/illumina_fastq/B71_example.illumina.R1.pair.fq.gz input/illumina_fastq/B71_example.illumina.R2.pair.fq.gz |
The ref.fasta contains the sequence of the region chr5:1000000–1050000 of B71Ref1.5.
| input/ref.fasta |
Procedure
Category
Plant Science > Plant molecular biology > DNA > DNA sequencing
Bioinformatics and Computational Biology
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.