
- Home
- Protocols

-

An Analysis Pipeline for Identification of RNA Modification, Alternative Splicing and Polyadenylation Using Third Generation Sequencing
*Contributed equally to this work Published: Jun 5, 2022 DOI: 10.21769/BioProtoc.4433 Views: 1395
Edited by: Jinfeng Chen Reviewed by: Zhengrong YuanChristian Sailer
Abstract
Nanopore sequencing based on Oxford Nanopore Technologies (ONT) and Pacific BioSciences (PacBio) single-molecule real-time (SMRT) long-read isoform sequencing (Iso-Seq) have shown great potential in detecting post-transcriptional regulation. Direct RNA sequencing (DRS) has the advantages in capturing RNA modification due to without PCR amplification which is the limitation of the next-generation sequencing (NGS). Here, we provide a comprehensive computational procedure for the quantification of RNA modification in single-base resolution based on DRS data. Moreover, we also provide procedure on the identification of alternative splicing (AS) and alternative polyadenylation (APA) based on both DRS and PacBio Iso-Seq data. The entire step was based on two packages (Nanom6A and PRAPI), which were based on Python language on Linux system.
Graphical abstract:
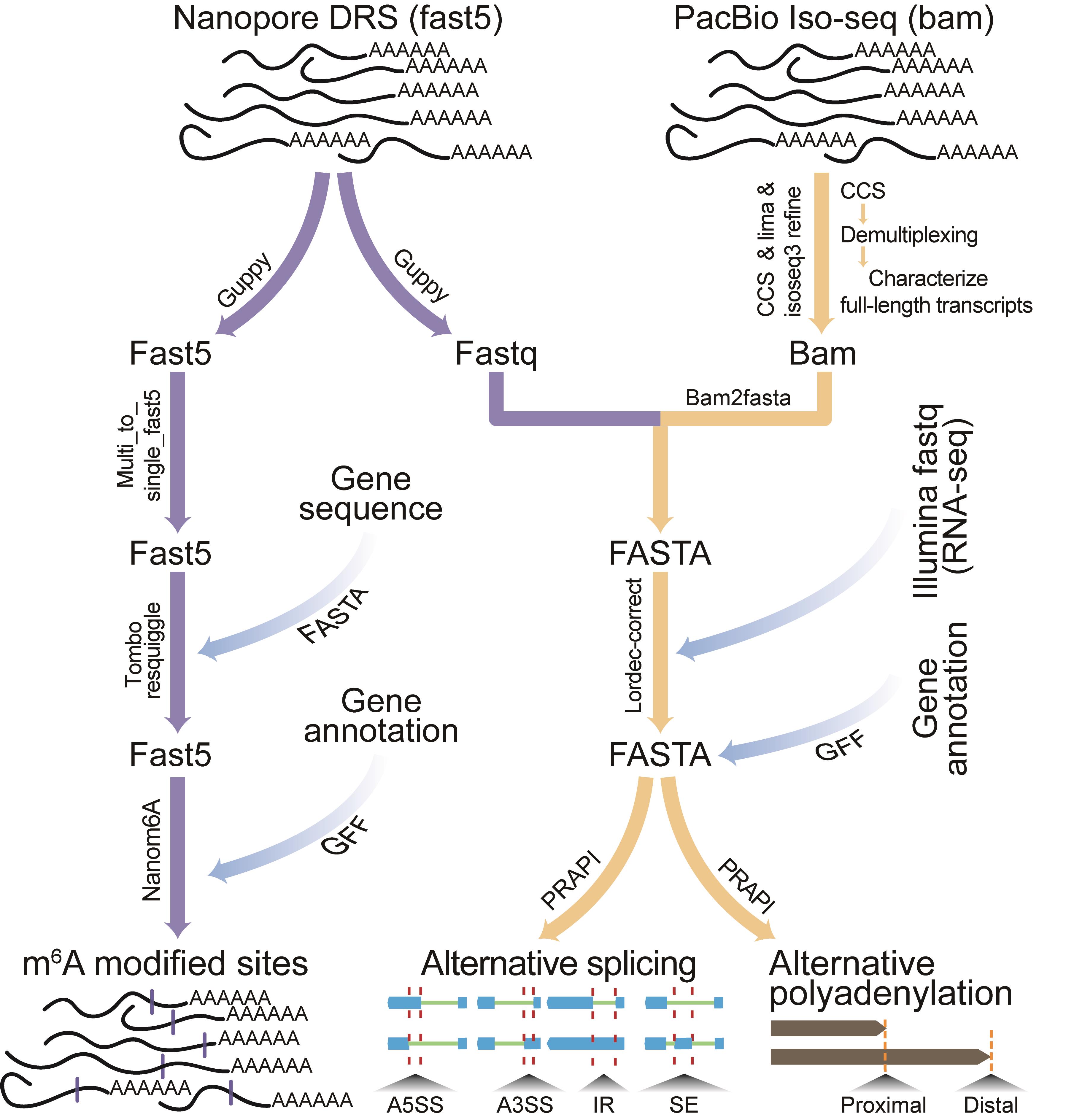
The flowchart for identification of RNA modification, alternative splicing and alternative polyadenylation based on third generation sequencing technology.
Background
Transcriptome provides an in-depth perspective for understanding the regulation in gene expression and growth development. With the rapid development of the third-generation sequencing technology, the data based on long-read sequencing presented great advantage in revealing post-transcriptional regulation (Smith et al., 2019). Post-transcription regulation has been known as a complex biological process, playing an important part in influencing the splicing, export, localization and translation of RNA (Keene, 2007). For example, alternative polyadenylation (APA) plays a vital role in maintaining the structure and stability of RNA (Di Giammartino et al., 2011, Tian and Manley, 2017). Alternative splicing (AS) is implicated in the regulation of rhizome-associated development (Wang et al., 2017) and hormone response (Zhang et al., 2018). A variety of AS events could be found in key genes related to biosynthesis pathways (Chen et al., 2020). PacBio Iso-Seq can capture the full-length mRNA in isoform-wide to analyze the post-transcriptional processing events such as AS and APA (Rhoads and Au, 2015). Accuracy and reliability tools are important to identify post-transcriptional events. AS based on short-read platform can be identified by several excellent software, such as rMATS (Shen et al., 2014) and SUPPA2 (Trincado et al., 2018). However short-read platforms show disadvantage in detecting full-length isoforms due to the limitation of transcriptome assembly (Rhoads and Au, 2015). TAPIS (Abdel-Ghany et al., 2016) and FLAIR (Tang et al., 2020) have been developed to identify AS events based on long-read sequencing. However, TAPIS does not provide quantitative analysis of post-transcriptional events (Abdel-Ghany et al., 2016). FLAIR can detect isoforms, but lacks APA detecting module (Tang et al., 2020). Hence, we provide a comprehensive and user-friendly pipeline (PRAPI) for the identification and visualization of AS and APA (Gao et al., 2018). Here we provide detail procedure for data processing, visualization, identification of AS and APA using PRAPI based on Iso-Seq isoforms and short read data.
In addition to PacBio platform, Oxford Nanopore Technologies (ONT) platform applies a unique measurement method to keep the information of RNA molecular passing through a special pore as an electrical signal (Garalde et al., 2018), which can identify RNA modifications including N6-methyladenosine (m6A) (Linder et al., 2015; Zhang et al., 2019), pseudouridine (Ψ) and 2′-O-methylation (Nm) (Begik et al., 2021) in single-nucleotide-resolution based on native RNA. Moreover, Nanopore DRS is capable of determining RNA secondary structure (Aw et al., 2021), and detecting amino acid from native unfolding protein sequence (Hu et al., 2021). Methods for detecting RNA modification based on ONT data have been reported in several software packages including Epinano (Liu et al., 2019), xPore (Pratanwanich et al., 2021), MINES (Lorenz et al., 2020), NanoDoc (Ueda, 2021), Nanom6A (Gao et al., 2021), Tombo (Stoiber et al., 2017). Epinano, xPore, Nanom6A, and MINES can achieve accuracy in single-base resolution. However, Epinano cannot distinguish m6A from other modified bases (such as m1A) due to depending on base-calling errors (Liu et al., 2019). Moreover, xPore requires m6A writer knockout lines as comparison and may be biased due to its strong enrichment in DRACH motif (Pratanwanich et al., 2021). Finally, MINES only detects four RRACH motifs (AGACT, GGACA, GGACC, and GGACT) and overlooked other 8 RRACH motifs (Lorenz et al., 2020). Nanom6A provides method using Nanopore direct RNA reads for identification of both qualitative and quantitative m6A modification in single-base resolution based on XGBoost algorithm, providing a highly precise transcriptome-wide identification, quantification and sequence contexts (RRACH) of m6A modification (Gao et al., 2021). In this study, we provide detail procedure for high accuracy of quantitation of RNA modification in single-base resolution. In brief, we provide comprehensive procedure on the identification of AS, APA and m6A based on DRS or PacBio Iso-Seq data. The Linux Bash Shells and scripts for Nanom6A and PRAPI analysis are now available in https://github.com/GuInNGS/NanoPrapi and https://github.com/Bio-protocol/Pipeline_for_Identification_m6A_AS_and_APA_with_Long_Read.
Equipment
Linux cluster (We recommend computer with at least 16GB RAM and multiple CPU cores)
Intensive computing is required for Guppy and Tombo which used fast5 data as input file
Server with Linux system (We recommend at least 16GB RAM)
Nanopore Sequencing
MinION or GridION Flow Cell (R9.4.1) (Oxford Nanopore Technologies, Cat. no. FLO-MIN106) (https://store.nanoporetech.com/flow-cell-r9-4-1.html)
Software and Data sets
Software
Anaconda (https://www.anaconda.com/products/individual)
A toolkit included thousands of open-source packages and libraries. User can also use Miniconda (https://docs.conda.io/en/latest/miniconda.html), a free minimal installer for Anaconda, to create environment and install software.
Guppy (v3.6.1) (https://community.nanoporetech.com/downloads)
Guppy from Oxford Nanopore sequencing data processing toolkit can perform basecalling to generate FASTQ file and an additional FAST5 file that contains basecalling information, which is available to ONT customers. Users should be an existing customer or register an account through the Nanopore community to download Guppy.
Ont_fast5_api (v0.3.2) (https://github.com/nanoporetech/ont_fast5_api)
Module of multi_to_single_fast5 was a conversion tool from Ont_fast5_api package to transfer single fast5 with big size into small size.
Tombo (Stoiber et al., 2017) (v1.5.1) (https://github.com/nanoporetech/tombo)
The re-squiggle algorithm from Tombo program assigned raw signal and associated base calls to transcriptome reference sequences for downstream analysis.
Nanom6A (Gao et al., 2021) (2021_3_18 version) (https://github.com/gaoyubang/nanom6A)
A software for identification and quantification of m6A modification at single-base-resolution base on Nanopore raw data.
PRAPI (Gao et al., 2018) (v1.0) (http://forestry.fafu.edu.cn/tool/PRAPI/)
Iso-Seq reads analysis tool (post-transcriptional regulation analysis pipeline), for identification of post-transcriptional events including AS and APA et. al.
LoRDEC (Salmela and Rivals, 2014) (v0.9) (https://gite.lirmm.fr/lordec/lordec-releases/-/wikis/home)
A tool for error correction of long reads from the third-generation sequencing using the short-sequence reads from second-generation sequencing platform. LoRDEC uses high-accuracy NGS data to construct de Bruijn Graph (DBG) for correcting the errors of the third-generation long reads from PacBio or Oxford platforms.
Picard (v2.26.8) (https://github.com/broadinstitute/picard)
GMAP (Wu and Watanabe, 2005; Wu and Nacu, 2010) (version 2019-12-01) (http://research-pub.gene.com/gmap/)
Input data
For m6A identification based on nanom6A, input data should be transcriptome reference sequences and the fast5 file from Nanopore DRS, which included raw signal. Each multi-fast5 file contains 4000 single fast5 reads. Each single fast5 read contains raw signal, and configuration information. The fast5 file is stored in HDF5 format and can be viewed by HDFView (https://www.hdfgroup.org/downloads/hdfview/). One typical HDF5 format looks like this (Figure 1).
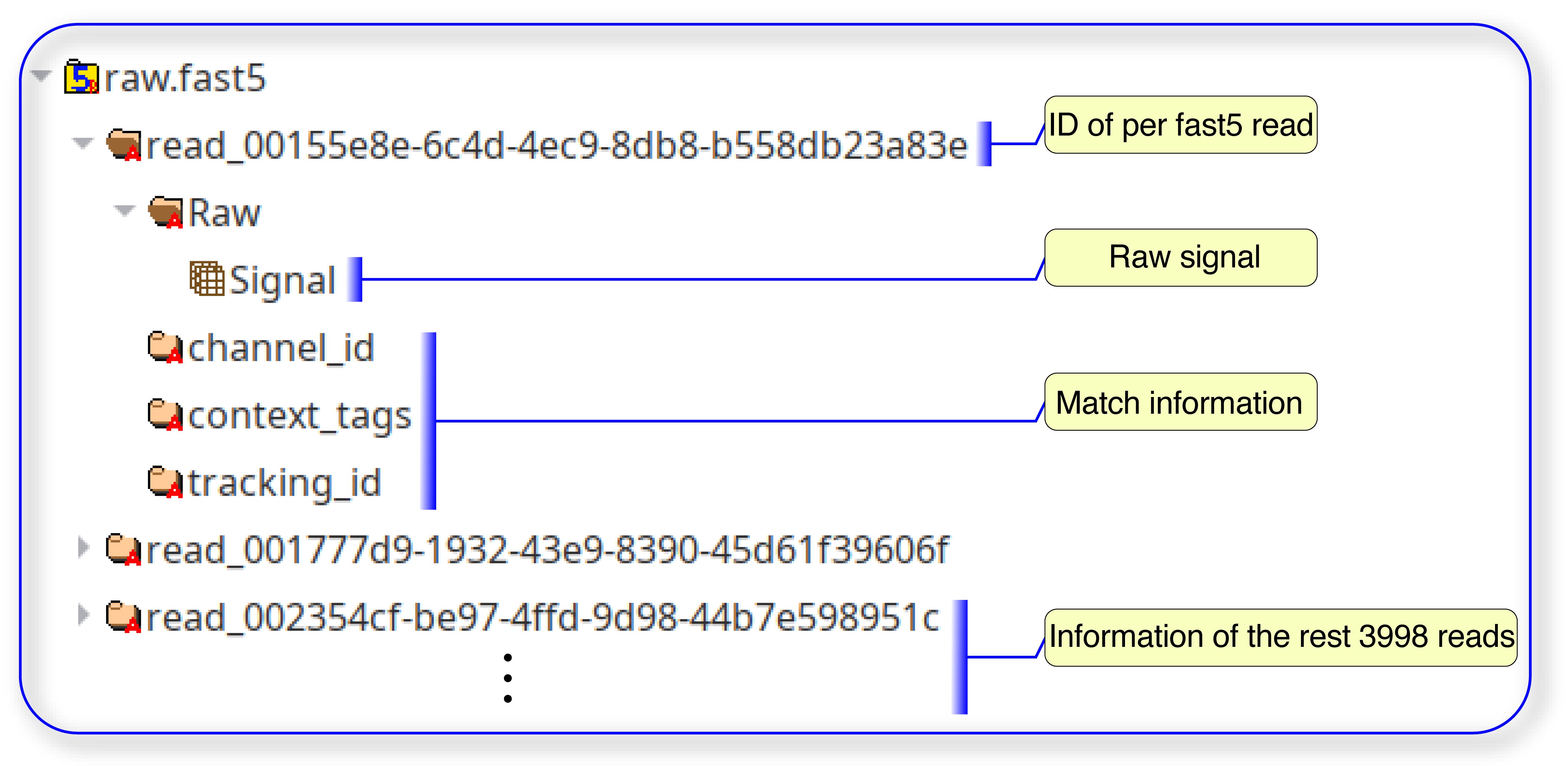
Figure 1. A typical HDF5 format for FAST5 file from DRS.In the example for identification of AS and APA based on PRAPI, input file should include genome file(.fasta), annotation file (genePred format), PacBio Iso-Seq read (.fasta) or Nanopore long reads (.fasta), which has been corrected by LoRDEC (Salmela and Rivals, 2014). PRAPI also can take RNA-Seq based on Illumina platform as input using sorted and indexed alignment files (BAM). The annotation file in GenePred table format (https://genome.ucsc.edu/FAQ/FAQformat.html#format9) is used during the process of AS identification. Annotation file with genePred format should have nine columns including name of gene, name of chromosome, strand, transcription start position, transcription end position, coding region start position, coding region end position, number of exons, exon start positions, and exon end positions, respectively.
Procedure
Category
Bioinformatics and Computational Biology
Plant Science > Plant molecular biology > RNA
Biological Engineering
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.