
- Home
- Protocols

-

Computational Analysis of Maize Enhancer Regulatory Elements Using ATAC-STARR-seq
Published: Mar 5, 2024 DOI: 10.21769/BioProtoc.4953 Views: 495
Reviewed by: G. Alex MasonPrashanth N Suravajhala
Abstract
The blueprints for development, response to the environment, and cellularfunction are largely the manifestation of distinct gene expression programscontrolled by the spatiotemporal activity of cis-regulatory elements. Althoughbiochemical methods for identifying accessible chromatin—a hallmark ofactive cis-regulatory elements—have been developed, approaches capable ofmeasuring and quantifying cis-regulatory activity are only beginning to berealized. Massively parallel reporter assays coupled to chromatin accessibilityprofiling present a high-throughput solution for testing thetranscription-activating capacity of millions of putatively regulatory DNAsequences in parallel. However, clear computational pipelines for analyzingthese high-throughput sequencing-based reporter assays are lacking. In thisprotocol, I layout and rationalize a computational framework for the processingand analysis of the transposase accessible chromatin profiling followed byself-transcribed active regulatory region sequencing (ATAC-STARR-seq) data froma recent study in Zea mays. The approach described herein canbe adapted to other sequencing-based reporter assays and is largely modelorganism–agnostic with appropriate input substitutions.
Keywords: STARR-seqBackground
Eukaryotic cells exhibit remarkable functional and morphological diversity despite containing a generally invariant copy of the same genomic sequence. Cellular heterogeneity arises in part due to the activities of cis-regulatory elements (CREs), short DNA-binding motifs recognized by sequence-specific transcription factors (TFs). CREs are often found in clusters termed cis-regulatory modules (CRMs) that dictate highly dynamic spatiotemporal patterns of gene expression via the cooperative activities of DNA-bound TFs (Schmitz et al., 2022). For proper activation of transcription, the cell strictly regulates CRM activity by controlling TF access of CRM sequences through nucleosome dynamics. Genome-wide approaches, such as the assay for transposase accessible chromatin sequencing (ATAC-seq), have been developed to profile accessible chromatin regions (ACRs) (Buenrostro et al., 2013; Minnoye et al., 2021). In general, CRMs that localize to accessible chromatin reflect active regulatory elements (Marand et al., 2017; Schmitz et al., 2022). Thus, activation and silencing of gene expression is effectively controlled by the relative chromatin accessibility of cognate CRMs.
CREs can be classified into distinct functional groups based on their regulatory effect on transcription, including enhancers, silencers, promoters, and insulators (Schmitz et al., 2022). Of these, enhancers are of particular interest due to their transcription-activating properties that function independently of location and orientation of their target genes, in contrast to the stereotypical locations of promoters surrounding gene transcription start sites (TSSs) (Marand et al., 2017; Schmitz et al., 2022). While analysis of chromatin accessibility in distinct tissues and cell types has been central to the identification of CRMs (Marand et al., 2021), chromatin profiling techniques are largely qualitative and lack the ability to quantitatively estimate regulatory activity. To overcome these challenges, massively parallel reporter assays (MPRAs) have been developed to quantify the transcription-activating properties of diverse sequences (Melnikov et al., 2012; Arnold et al., 2013). In particular, self-transcribing active regulatory region sequencing (STARR-seq) demonstrates the greatest potential for broad application by eliminating the need for homogenous cell lines available only in mammalian models typical of other MPRA methods (Arnold et al., 2013; Ricci et al., 2019; Sun et al., 2019; Jores et al., 2020). Although STARR-seq was originally designed to profile the entire genome for regulatory activity, recent implementations have successfully utilized ATAC-seq libraries as input (ATAC-STARR-seq), reducing the search space to potential regulatory regions and offsetting sequencing costs and library complexity requirements (Figure 1). Despite its promise as a powerful approach towards understanding cis-regulatory activity, computational analysis of ATAC-STARR-seq data remains challenging, particularly due to a lack of dedicated software and computational pipelines.
Here, I present a computational pipeline for analysis of ATAC-STARR-seq data generated in Zea mays L., cultivar B73 (Ricci et al., 2019). After processing and evaluation of data quality, I demonstrate how ATAC-STARR-seq data analysis allows for the interrogation of new biological questions. The pipeline can be run entirely from the code below or through freely available bash, perl, and R scripts hosted at https://github.com/Bio-protocol/Maize_ATAC_STARR_seq.
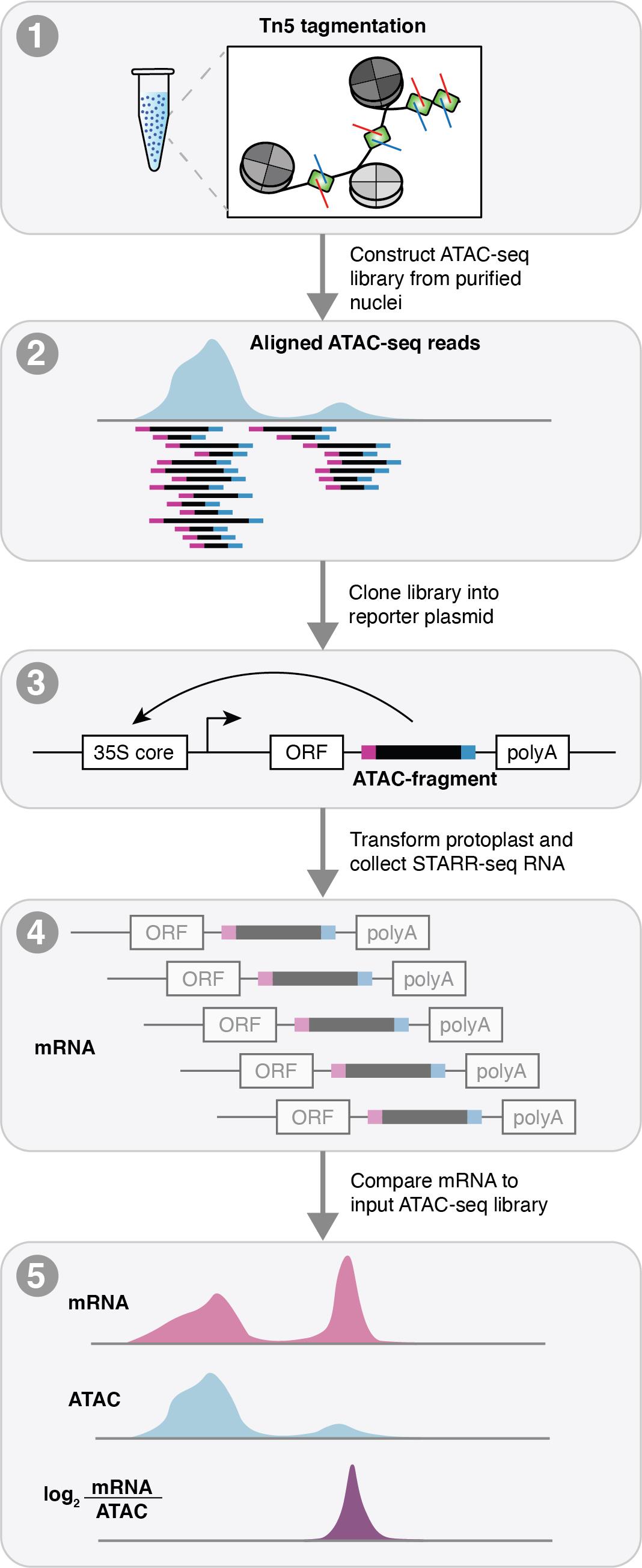
Figure 1. Schematic of assay for transposase accessible chromatin profiling followed by self-transcribed active regulatory region sequencing (ATAC-STARR-seq). ATAC-STARR-seq begins by first generating an ATAC-seq library. The ATAC fragments are then cloned into a reporter assay and transformed into maize protoplasts. Transformed protoplasts are then split into two pools: the first for sequencing the input fragments (ATAC-seq DNA) and the second for purifying transcribed (mRNA) ATAC-seq fragments that facilitate their own transcription from the reporter construct. Raw sequenced reads for the ATAC-seq input and mRNA output are processed and aligned to the maize reference genome and compared to provide estimates of cis-regulatory activity.
Equipment
This pipeline assumes that a user has knowledge of shell commands and is comfortable working on a Linux-based operating system.
Computational requirements
The following procedure can be run on any Linux-like system. However, this protocol and publicly available code is written for executing commands via a high-performance computing (HPC) cluster managed by a SLURM scheduler. Still, the code presented here can be readily converted to TORQUE or other HPC systems. The pipeline assumes a working Perl interpreter version 5.30.0 or greater and R version 3.6.2 or greater.
Software
The following analytical procedure makes use of several standard computational tools that are assumed to be available in the user’s shell environment:
BWA MEM (Li and Durbin, 2009) v0.7.17; http://bio-bwa.sourceforge.net/bwa.shtml
SAMtools (Li et al., 2009) v1.14; http://www.htslib.org
BEDtools (Quinlan and Hall, 2010) v2.27.1; https://bedtools.readthedocs.io/en/latest/
SRA-toolkit (Leinonen et al., 2011) v2.11.1; https://github.com/ncbi/sra-tools
fastp (Chen et al., 2018) v0.20.0; https://github.com/OpenGene/fastp
pigz v2.4; https://zlib.net/pigz/
MACS2 (Liu, 2014) v2.2.7.1; https://pypi.org/project/MACS2/
UCSC binaries (Kent et al., 2010) v1.04.0; http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/
tabix (Li, 2011) v0.2.6; http://www.htslib.org/doc/tabix.html
IGV (Thorvaldsdottir et al., 2013) v2.11.1; https://software.broadinstitute.org/software/igv
MEME (Grant et al., 2011) v5.4.1; https://meme-suite.org/meme/index.html
CrossMap (Zhao et al., 2014) v0.5.1; http://crossmap.sourceforge.net/
DeepTools (Ramirez et al., 2014) v3.5.1; https://deeptools.readthedocs.io/en/develop/index.html
Input data
The starting input for this computational pipeline uses paired-end sequencing data from an ATAC-STARR-seq experiment performed on maize protoplasts (Ricci et al., 2019). The ATAC-STARR-seq experiment consisted of a DNA input (ATAC-seq library) and a mRNA readout (self-transcribed regulatory regions) to identify genomic regions exhibiting transcription-activating regulatory activity.
Transfected ATAC-seq DNA-input FASTQ
Transcribed ATAC-seq mRNA FASTQ
Procedure
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
Category
Bioinformatics and Computational Biology
Plant Science > Plant molecular biology > Genetic analysis
Systems Biology > Genomics > Functional genomics
Do you have any questions about this protocol?
Post your question to gather feedback from the community. We will also invite the authors of this article to respond.