

A Computational Workflow for Membrane Protein–Ligand Interaction Studies: Focus on α5-Containing GABA (A) Receptors
膜蛋白–配体相互作用研究的计算流程:以含 α5 亚基的 GABA(A) 受体为例

发布: 2025年11月20日第15卷第22期 DOI: 10.21769/BioProtoc.5515 浏览次数: 2520
评审: Anonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Oct 2025
Advertisement










Abstract
In neuropharmacology and drug development, in silico methods have become increasingly vital, particularly for studying receptor–ligand interactions at the molecular level. Membrane proteins such as GABA (A) receptors play a central role in neuronal signaling and are key targets for therapeutic intervention. While experimental techniques like electrophysiology and radioligand binding provide valuable functional data, they often fall short in resolving the structural complexity of membrane proteins and can be time-consuming, costly, and inaccessible in many research settings. This study presents a comprehensive computational workflow for investigating membrane protein–ligand interactions, demonstrated using the GABA (A) receptor α5β2γ2 subtype and mitragynine, an alkaloid from Mitragyna speciosa (Kratom), as a case study. The protocol includes homology modeling of the receptor based on a high-resolution template, followed by structure optimization and validation. Ligand docking is then used to predict binding sites and affinities at known modulatory interfaces. Finally, molecular dynamics (MD) simulations assess the stability and conformational dynamics of receptor–ligand complexes over time. Overall, this workflow offers a robust, reproducible approach for structural analysis of membrane protein–ligand interactions, supporting early-stage drug discovery and mechanistic studies across diverse membrane protein targets.
Key features
• Applicable to diverse membrane proteins, including ion channels and G protein-coupled receptors (GPCRs), facilitating ligand interactions and dynamic behavior in biologically relevant environments.
• Supports the investigation of both natural and synthetic compounds targeting specific receptor subtypes within complex membrane systems.
• Combines homology modeling, molecular docking, and molecular dynamics simulations to deliver comprehensive structural and functional insights.
• Showcased with the GABA (A) α5β2γ2 receptor subtype and alkaloid from Mitragyna speciosa, with adaptability to a broad range of receptor–ligand systems.
Keywords: Membrane protein modeling (膜蛋白建模)Graphical overview
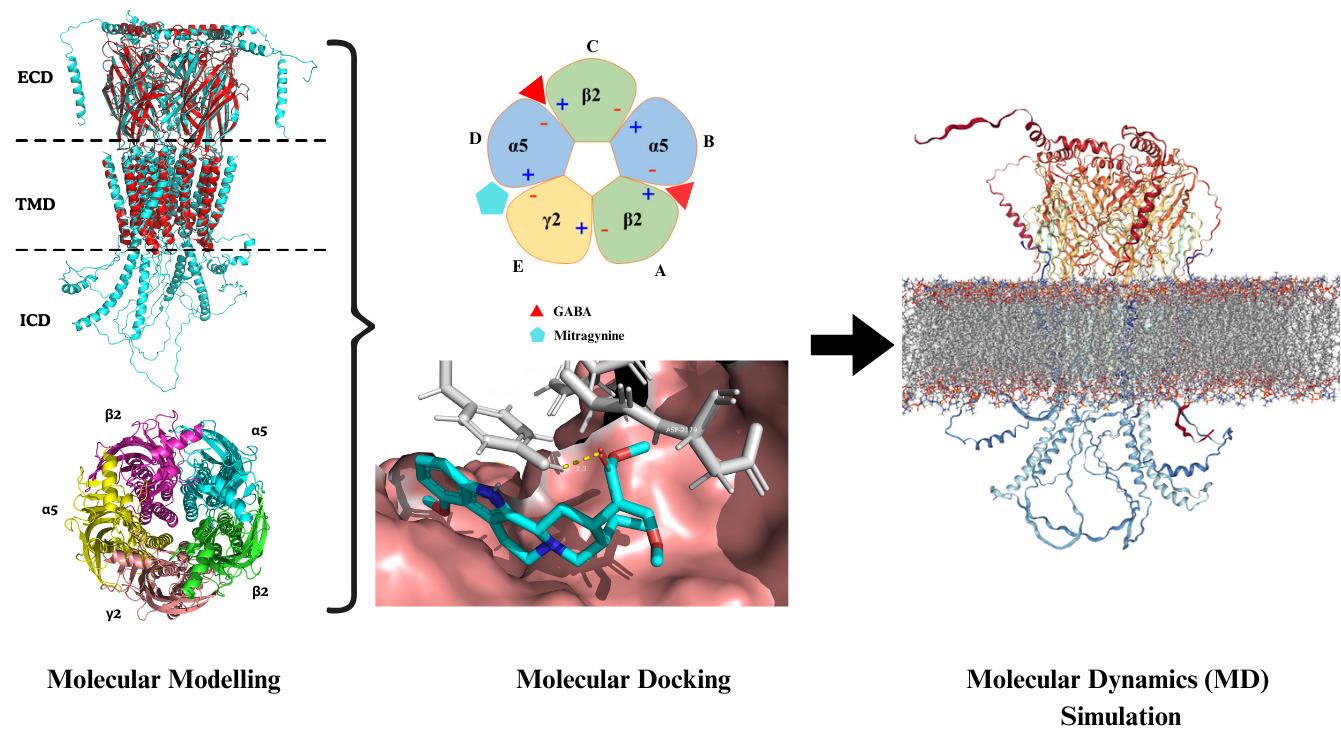
Comprehensive in silico methodological framework designed to investigate the molecular interactions between the alkaloid mitragynine and the α5β2γ2 subtype of the hippocampal GABA (A) receptor
Background
In the mammalian central nervous system (CNS), γ-aminobutyric acid (GABA) is the primary inhibitory neurotransmitter [1]. GABA (A) receptors are ionotropic receptors that, upon GABA binding, open chloride (Cl-) channels, leading to neuronal hyperpolarization and inhibition [2]. These receptors are pentameric ligand-gated ion channels, typically assembled from five subunits selected from 19 known types: α1–6, β1–3, γ1–3, δ, ε, θ, π, and ρ1–3 [3]. The most common brain configuration consists of two α, two β, and one additional subunit, forming αβγ or αβδ combinations, which correspond to synaptic and extrasynaptic receptors, respectively [4]. The specific subunit composition determines each receptor's pharmacological profile, regional brain distribution, and physiological role. Among the subunits, α5 is particularly notable for its involvement in cognitive processes, including learning and spatial memory [5–7]. The α5β2γ2 GABA (A) receptor subtype has been identified as a key modulator of cognition and anxiety-related behaviors. This receptor subtype is highly expressed in the hippocampus, particularly in the CA1 and CA3 regions, areas critical for memory encoding and spatial navigation [8,9]. Its anatomical localization and functional role make α5β2γ2 a compelling target for cognitive modulation. In this context, natural compounds with neuroactive properties may offer new avenues for modulating GABAergic signaling.
Mitragyna speciosa (commonly known as Kratom) is a tropical medicinal plant traditionally used for its stimulant and opioid-like effects [10]. Among its constituents, mitragynine is the primary and pharmacologically active alkaloid. Mitragynine exhibits analgesic, anti-inflammatory, and psychoactive properties [11], primarily acting as a partial agonist at the μ-opioid receptor and an antagonist at κ- and δ-opioid receptors [12]. Beyond its opioid-related activity, mitragynine has shown potential to interact with receptors involved in cognitive processes, raising interest in its neuromodulatory effects on GABAergic systems [13]. To systematically explore this potential, we developed an integrated in silico workflow to investigate the interaction of mitragynine with the α5β2γ2 GABA (A) receptor subtype. The protocol begins with homology modeling of the receptor using the α1β2γ2 subtype as a structural template. Following model optimization and validation, molecular docking is performed to predict mitragynine’s binding affinity and identify key interaction sites. Finally, molecular dynamics (MD) simulations are used to assess the stability and conformational behavior of the receptor–ligand complex. Overall, this computational approach provides detailed structural and dynamic insights into the potential neuromodulatory action of mitragynine on a cognitively significant GABA (A) receptor subtype.
Equipment
Hardware
For molecular docking and MD simulations, especially in in silico studies, access to adequate computational resources is essential for efficient performance. While the type or number of computing devices does not inherently affect the scientific validity or reproducibility of the results when using deterministic algorithms with identical parameters, it significantly impacts processing speed and overall throughput. In particular, parallel processing capabilities can greatly reduce simulation time and enhance workflow scalability. Suggested hardware specifications for an optimal workstation setup are summarized in Table 1.
Table 1. Specification summary of computing systems used in this study
| Specification | High-performance computer | Workstation custom PC |
| Model | Gigabyte Technology Co., Ltd. Z790 UD AX | Gigabyte Technology Co., Ltd. B550 AOROS ELITE |
| Processor type | Intel® CoreTM i9-14900K × 32 | AMD® Ryzen 5 5600x 6-core processor × 12 |
| Memory | 32 GB RAM | 32 GB RAM |
| Graphic processing unit (GPU) | NVIDIA GeForce RTX 4080/PCIe/SSE2/NVIDIA Corporation | NVIDIA Corporation GA106 [GeForce RTX 3060 Lite Hash Rate] |
| Disk capacity | 5.9 TB | 2.5 TB |
| Operating system | Ubuntu 22.04.5 LTS | Ubuntu 22.04.3 LTS |
Software and datasets
Software
This section provides an overview of the key software packages employed in the study, including molecular modeling platforms, simulation engines, and visualization tools, as summarized in Table 2.
Table 2. Summary of computational tools employed and their functional roles
| Software | Version | Function | References |
PyMOL | 2.5 | To display protein and ligand structures in 3D To allow molecular editing and measure distances, angles, and torsion between atoms and residues To render high-quality molecular graphics | [14] |
| MODELLER | 10.7 | To add missing residues To align the target sequences To generate 3D models of proteins by using the homologous structure as the template To rank and validate the quality of predicted models via the DOPE score | [15] |
| GROMACS | 2023.4 | To perform energy minimization and system equilibration To simulate protein complexes over time To analyze MD output | [16] |
| AutoDock Tools | 4.2 | To prepare ligand topology To assign partial charges to the protein To define a grid box over the target binding site | [17] |
| AutoDock Vina | 1.2.0 | To predict the binding poses and affinities of ligands To generate output files in PDBQT format | [18] |
| GRACE | 5.1.25 | To generate graphs To support overlaying multiple datasets for comparison | [19] |
| VMD | 1.9.4 | To load and playback MD simulation trajectories To generate high-quality molecular images and videos | [20] |
| Discovery Studio | 2021 | To analyze and visualize protein-ligand interactions in 2D and 3D | [21] |
Database and repositories
This section provides the validated and structured biological information required for each feature, as summarized in Table 3.
Table 3. Resources and databases utilized in structural and molecular analysis
| Name | Feature | URL |
| RCSB Protein Database (RCSB PDB) | Database for the three-dimensional structural data of large biological molecules such as proteins and nucleic acids | https://www.rcsb.org/ |
| UniProt | Database for protein sequences | https://www.uniprot.org/ |
| AlphaFold | Program for protein structure prediction | https://alphafold.ebi.ac.uk/ |
| Clustal Omega | Multiple sequence alignment programs | https://www.ebi.ac.uk/jdispatcher/msa/clustalo |
| Phylip to Pir sequence converter | Program for translation of the sequence alignment data between two formats | https://sequenceconversion.bugaco.com/converter/biology/sequences/pir_to_phylip.php |
| UCLA-DOE LAB | Verification of protein structures | https://saves.mbi.ucla.edu/ |
| CHARMM-GUI | Constructing coarse-grained, atomistic models, suitable for simulation, with a single or complex interest in solution or membrane settings | https://www.charmm-gui.org/ |
| PubChem | Database for chemical molecules | https://pubchem.ncbi.nlm.nih.gov/ |
| Protein Ligand Interaction Profiler (PLIP) | Identification of non-covalent interactions between biological macromolecules and ligands | https://plip-tool.biotec.tu-dresden.de/plip-web/plip/index |
File organization and directory setup
This section provides the structured directory information required across different stages of the in silico pipeline, as illustrated in Figure 1. Establishing a systematic folder hierarchy ensures proper file management, reproducibility, and efficient access to input, output, and analysis files.
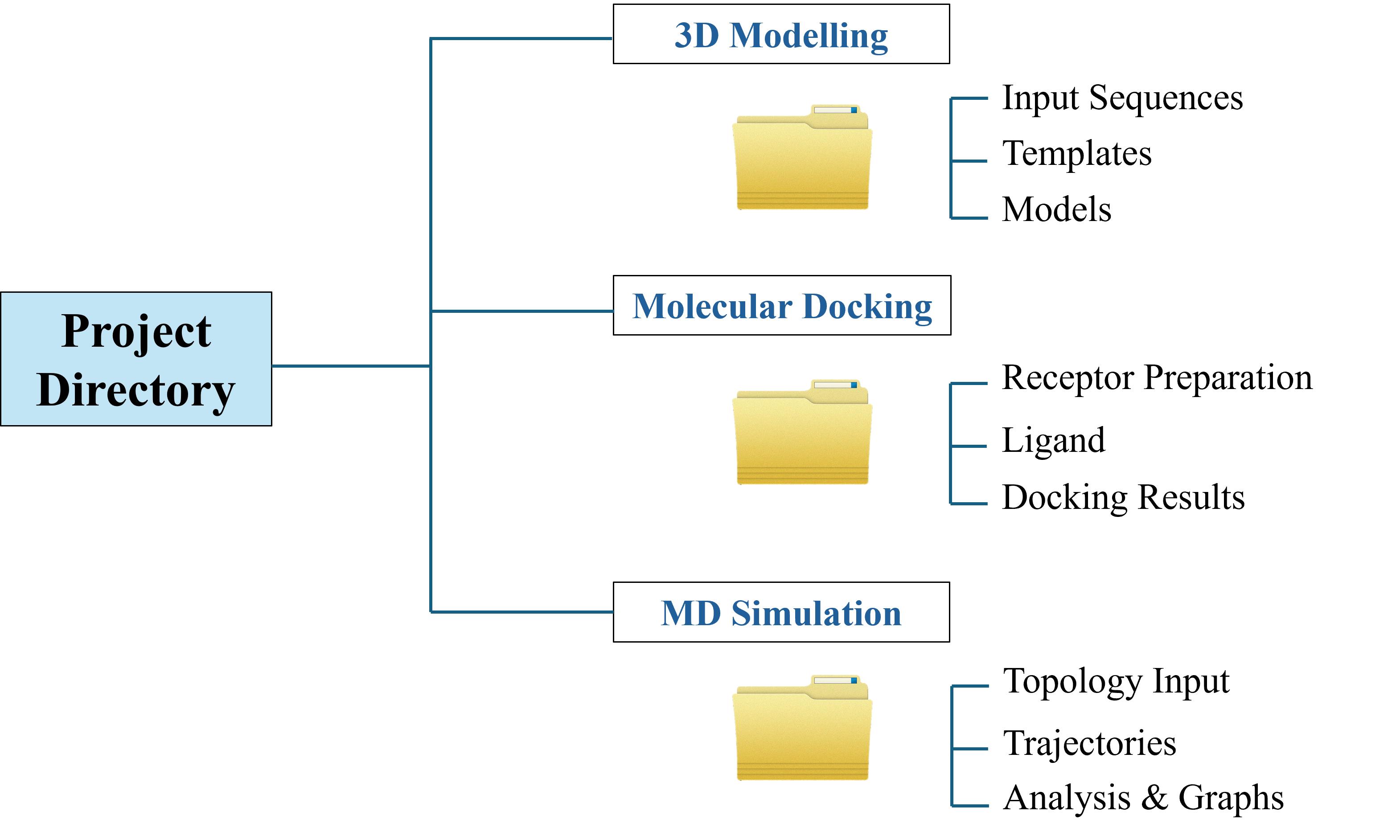
Figure 1. Directory structure for the in silico workflow
Procedure
文章信息
稿件历史记录
提交日期: Aug 20, 2025
接收日期: Oct 14, 2025
在线发布日期: Oct 30, 2025
出版日期: Nov 20, 2025
版权信息
© 2025 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
如何引用
Mohamad, S. M. S., Halim, K. B. A., Hamid, A. A. A. and Has, A. T. C. (2025). A Computational Workflow for Membrane Protein–Ligand Interaction Studies: Focus on α5-Containing GABA (A) Receptors. Bio-protocol 15(22): e5515. DOI: 10.21769/BioProtoc.5515.
分类
生物信息学与计算生物学
生物化学 > 蛋白质 > 相互作用 > 蛋白质-配体相互作用
系统生物学 > 相互作用组 >
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。