

A Comprehensive Protocol for Bayesian Phylogenetic Analysis Using MrBayes: From Sequence Alignment to Model Selection and Phylogenetic Inference
基于MrBayes的贝叶斯系统发育分析全流程方案:从序列比对到模型选择与系统发育推断
发布: 2025年04月20日第15卷第8期 DOI: 10.21769/BioProtoc.5276 浏览次数: 2487
评审: Prashanth N SuravajhalaSuresh PantheeAnonymous reviewer(s)
Advertisement










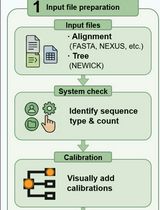
Abstract
Bayesian phylogenetic analysis is essential for elucidating evolutionary relationships among organisms. Traditional methods often rely on fixed models and manual parameter settings, which can limit accuracy and efficiency. This protocol presents an integrated workflow that leverages GUIDANCE2 for rigorous sequence alignment, ProtTest and MrModeltest for robust model selection, and MrBayes for phylogenetic tree estimation through Bayesian inference. By automating key steps and providing detailed command-line instructions, this protocol enhances the reliability and reproducibility of phylogenetic studies.
Key features
• Robust sequence alignment: Combines GUIDANCE2 and MAFFT to handle complex evolutionary events.
• Automated model selection: Utilizes ProtTest and MrModeltest for protein evolution models and nucleotide substitution models, respectively.
• Streamlined workflow: Provides step-by-step instructions from sequence alignment to phylogenetic tree estimation through Bayesian inference.
Keywords: Bayesian phylogenetic analysis (贝叶斯系统发育分析)Background
Phylogenetic analysis plays a critical role in understanding the evolutionary relationships among species, informing diverse fields such as evolutionary biology, epidemiology, and conservation genetics. The process of generating a phylogenetic tree typically involves key steps including sequence alignment, model selection, and tree inference, each of which is essential for deriving reliable evolutionary conclusions. However, traditional phylogenetic workflows often involve manual sequence alignment and model selection, introducing potential biases and inefficiencies.
To address these challenges, numerous computational tools have been developed. For example, GUIDANCE2 enhances sequence alignment by accounting for alignment uncertainty and evolutionary events such as insertions and deletions [1]. Model selection tools like Protest [2] and MrModeltest2 [3] automate the identification of optimal evolutionary models using statistical criteria such as AIC and BIC, thereby improving the reliability of downstream phylogenetic inferences. Besides, tools such as PAUP* [4] enable comprehensive phylogenetic analysis for nucleotide sequences, while MEGA X [5] facilitates sequence format conversion and preliminary analyses.
Beyond these tools, several non-Bayesian phylogenetic inference methods offer powerful alternatives with distinct advantages. The PHYLIP package [6] provides a comprehensive suite of programs implementing distance matrix, maximum parsimony, and maximum likelihood methods, making it a versatile choice for diverse phylogenetic analyses. Maximum likelihood-based programs like RAxML [7] and IQ-TREE [8] have revolutionized the field with their computational efficiency and accuracy, especially for large datasets. FastTree [9] employs heuristic approaches to construct approximately maximum-likelihood phylogenetic trees with remarkable speed while maintaining reasonable accuracy. PhyML [10] offers robust algorithms for maximum likelihood tree estimation with extensive substitution model options and branch support assessment. These non-Bayesian tools provide complementary strengths to Bayesian methods, often excelling in computational efficiency while still delivering statistically sound phylogenetic inferences.
Bayesian methods, particularly those implemented in MrBayes [11], provide a robust probabilistic framework for estimating phylogenetic trees and evolutionary parameters by incorporating uncertainty and prior knowledge. However, integrating these tools into a cohesive and reproducible workflow remains challenging due to differing format requirements between tools. For example, GUIDANCE2 accepts FASTA/PHYLIP inputs, MrBayes requires NEXUS format [12], and PAUP* demands non-interleaved NEXUS [4,13] for its analyses. These diverging specifications create hidden technical barriers. Our protocol addresses these challenges by presenting a seamless, step-by-step guide that integrates sequence alignment, model selection, and Bayesian inference using MrBayes. It automates critical steps, minimizes manual intervention, reduces potential errors, and ensures reproducibility. Custom Python scripts are included to streamline the parsing of model selection outputs, enhancing data handling efficiency. This structured protocol simplifies the phylogenetic analysis process, improves the accuracy and reliability of results, and is applicable to diverse datasets, including both protein and nucleotide sequences.
The NEXUS format is a common data format for phylogenetic analysis, facilitating greater cooperation in the analysis and visualization of data. PAUP* reads data in NEXUS file format, and all NEXUS files must begin with the declaration "#NEXUS". The Newick format [14] is another widely used format for representing phylogenetic trees, and it is supported by many phylogenetic analysis tools. The protocol leverages MEGA for initial format conversions and PAUP* for format refinement, ensuring seamless data handoffs between tools and preventing pipeline failures from format mismatches. This approach systematically addresses integration challenges and provides a versatile and reliable resource for researchers conducting rigorous evolutionary studies.
Software and datasets
All procedures in this protocol were developed and tested on Windows 10.
1. Python (Version: 3.13.1)
a. Homepage: https://www.python.org/
b. Downloads: https://www.python.org/ftp/python/3.13.1/python-3.13.1-amd64.exe
c. Platform: Windows
d. Last accessed: February 2025
e. Installation steps: Follow the instructions in the software installation package to install.
2. JAVA (Version: 8 or later)
a. Homepage: https://www.java.com/en/
b. Downloads: https://www.java.com/en/download/
c. Platform: Windows
d. Last accessed: February 2025
e. Installation steps: Follow the instructions in the software installation package to install.
3. PAUP* (Version: 4.0a Build 169)
a. Homepage: https://paup.phylosolutions.com/
b. Downloads: https://phylosolutions.com/paup-test/
c. Platform: Windows
d. Last accessed: February 2025
e. Installation steps: Follow the instructions in the software installation package to install.
4. MEGA X
a. Homepage: https://www.megasoftware.net/
b. Downloads: https://www.megasoftware.net/dload_win_gui
c. Platform: Windows
d. Last accessed: February 2025
e. Installation steps: Follow the instructions in the software installation package to install.
5. MrModeltest2 (Version: 2.4)
a. Homepage: https://github.com/nylander/MrModeltest2
b. Downloads: https://github.com/nylander/MrModeltest2/releases/tag/v.2.4
c. Platform: Windows, dependent on PAUP*
d. Last accessed: February 2025
e. No software installation is required: Copy the MrModelblock file from MrModelTest to your working directory, execute it in PAUP* via File > Execute, and use the generated mrmodel.scores file for subsequent analyses.
6. ProtTest (Version: 3.4.2)
a. Homepage: https://github.com/ddarriba/prottest3
b. Downloads: https://github.com/ddarriba/prottest3/releases/tag/3.4.2-release
c. Platform: Windows, Dependent on JAVA
d. Last accessed: February 2025
e. Installation steps: Download the latest ProtTest version (prottest-3.4.2-20160508.tar.gz) from its GitHub page and ensure Java is already installed. Extract the files to a directory with only English characters and no spaces. To use ProtTest, navigate to its extraction path in the command line terminal.
7. MrBayes (Version: 3.2.7a)
a. Homepage: https://nbisweden.github.io/MrBayes/
b. Downloads: https://github.com/NBISweden/MrBayes/releases/tag/v3.2.7
c. Platform: Windows
d. Last accessed: February 2025
e. Installation steps: Download MrBayes-3.2.7-WIN.zip from its GitHub page and extract it to a directory with only English characters and no spaces; inside the extracted bin folder, rename mb.3.2.7-win64.exe (for 64-bit CPUs) or mb.3.2.7-win32.exe (for 32-bit CPUs or if unsure) to mb.exe; place your NEXUS files in this directory, open a command line terminal here by holding the Shift key, right-clicking inside the folder, and selecting Open command window here (or Open PowerShell window here), then type mb and press Enter to launch MrBayes.
System requirements
To enhance the reproducibility and accessibility of this protocol, the following hardware specifications are recommended as minimal requirements for computational implementation:
1. Processor: A single-core central processing unit (CPU) with a base clock speed ≥2.0 GHz
2. Memory: 2 GB of random access memory (RAM)
3. Storage: 15 GB available disk space for software installations, intermediate files, and output storage
4. Graphics: No graphical processing unit (GPU) acceleration required
While these specifications suffice for basic analyses, multi-core processors (>4 cores) and expanded RAM (≥8 GB) are strongly recommended for improving computational efficiency during Bayesian inference with large datasets.
Procedure
文章信息
稿件历史记录
提交日期: Jan 4, 2025
接收日期: Mar 12, 2025
在线发布日期: Mar 26, 2025
出版日期: Apr 20, 2025
版权信息
© 2025 The Author(s); This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/).
如何引用
Wang, J., Chen, F., Xiao, X., Yang, X. and Xia, W. (2025). A Comprehensive Protocol for Bayesian Phylogenetic Analysis Using MrBayes: From Sequence Alignment to Model Selection and Phylogenetic Inference. Bio-protocol 15(8): e5276. DOI: 10.21769/BioProtoc.5276.
分类
生物信息学与计算生物学
系统生物学 > 基因组学 > 种系遗传学
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。