

Annotated Bioinformatic Pipelines for Phylogenomic Placement of Mitochondrial Genomes
线粒体基因组系统发育分析的生物信息学流程详解
发布: 2025年03月05日第15卷第5期 DOI: 10.21769/BioProtoc.5232 浏览次数: 2651
评审: Waldir Miron Berbel-FilhoPrashanth N SuravajhalaMaria Lynn Spletter
参见作者原研究论文
The authors used this protocol in:

Jan 2024
Advertisement










Abstract
The limited standards for the rigorous and objective use of mitochondrial genomes (mitogenomes) can lead to uncertainties regarding the phylogenetic relationships of taxa under varying evolutionary constraints. The mitogenome exhibits heterogeneity in base composition, and evolutionary rates may vary across different regions, which can cause empirical data to violate assumptions of the applied evolutionary models. Consequently, the unique evolutionary signatures of the dataset must be carefully evaluated before selecting an appropriate approach for phylogenomic inference. Here, we present the bioinformatic pipeline and code used to expand the mitogenome phylogeny of the order Carcharhiniformes (groundsharks), with a focus on houndsharks (Chondrichthyes: Triakidae). We present a rigorous approach for addressing difficult-to-resolve phylogenies, incorporating multi-species coalescent modelling (MSCM) to address gene/species tree discordance. The protocol describes carefully designed approaches for preparing alignments, partitioning datasets, assigning models of evolution, inferring phylogenies based on traditional site-homogenous concatenation approaches as well as under multispecies coalescent and site heterogenous models, and generating statistical data for comparison of different topological outcomes. The datasets required to run our analyses are available on GitHub and Dryad repositories.
Key features
• An extensive statistical framework to conduct model selection and data partitioning and tackle difficult-to-resolve phylogenies.
• Instructions for generating statistical data for comparison of different topological outcomes.
• Tips for selecting mitochondrial phylogenomic (mitophylogenomic) approaches to suit unique datasets.
• Access to the scripts, data files, and pipelines used to enable replication of all analyses.
Keywords: Taxonomy (分类学)Graphical overview
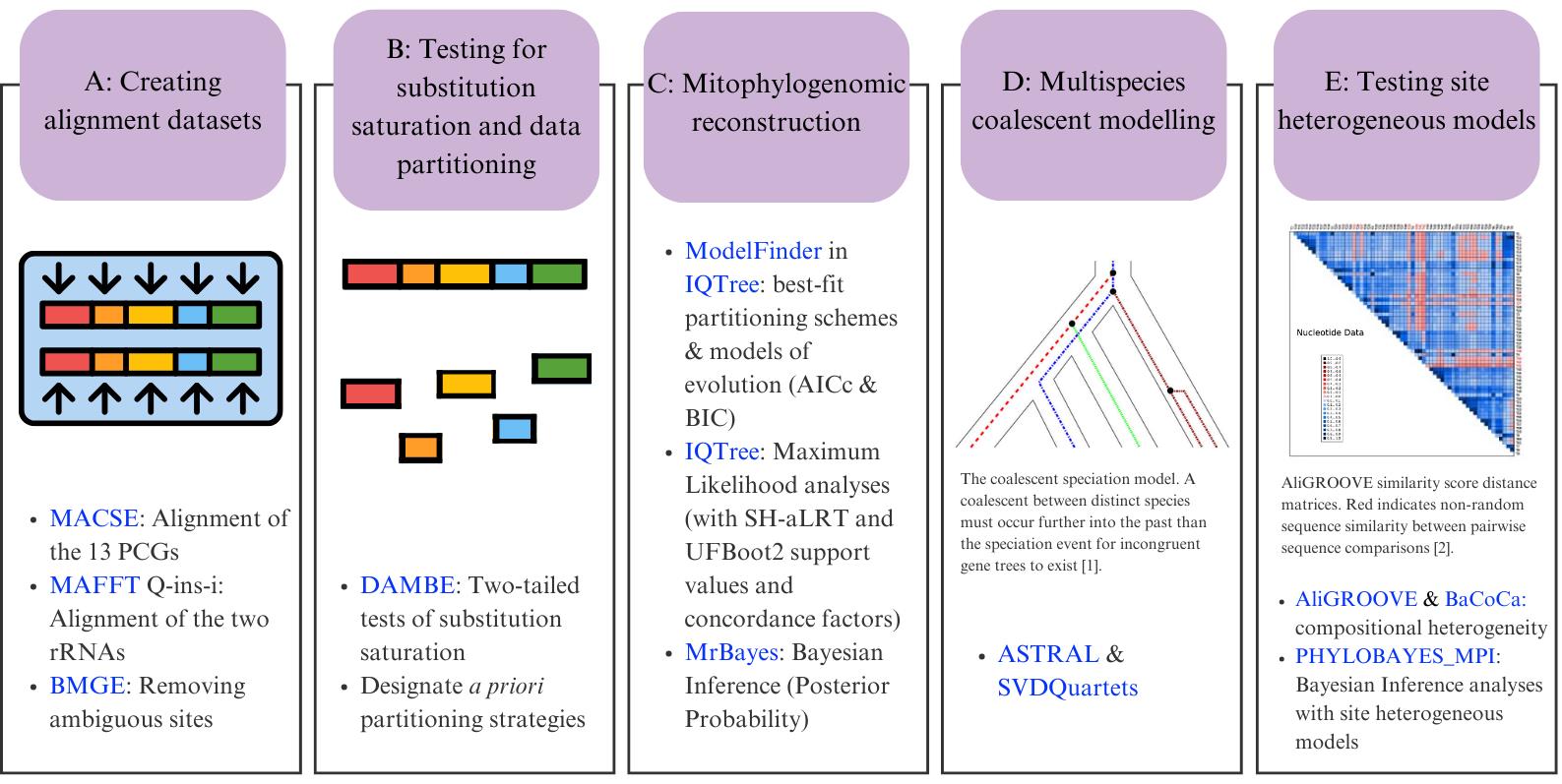
Bioinformatic workflow for phylogenomic reconstruction using mitochondrial genomes. In section D, the image shows the coalescent speciation model [1]; section E shows an example of sequence heterogeneity in pairwise sequence comparisons [2]. Software programs are indicated in blue.
Background
Phylogenetics, the study of the evolutionary relationship between organisms, has gained momentum following the genomics revolution, which has allowed for the accumulation of high-quality mitochondrial genomes (mitogenomes) [3–6]. However, even with larger datasets, we are still left with inconsistencies in the phylogenetic placement of many groups, from genera to orders [7,8]. Consequently, although it is vital to ensure taxa are adequately represented [9,10], phylogenetic analysis methods are often more integral to the interpretations of hypotheses than simply accumulating more data [7,11].
Traditional mitophylogenomic studies partition concatenated alignments based on sequence properties such as gene boundaries and codon locations before selecting and optimising substitution models for each partition [12–16]. There is heterogeneity in base composition and evolutionary rates at different scales across the mitogenome [17,18], so the empirical data may violate the assumptions of the applied models of evolution. Consequently, tools like ModelFinder have been exploited to select substitution models that best fit predefined subsets of a given dataset according to a chosen statistical criterion under a maximum likelihood (ML) framework [14,16,19]. However, the a priori partitioning strategy fed into the software is often selected without accounting for the unique evolutionary signatures of a dataset [14,16,20,21]. On the one hand, compositional heterogeneity, substitution saturation, branch length heterogeneity, and incomplete lineage sorting can lead to model violations and profoundly impact phylogenetic outcomes [22–26]. On the other hand, using phylogenetic strategies that account for these factors without first testing for their presence can also be detrimental to accurate tree construction. Overpartitioning the dataset or incorrectly using site-specific models can lead to “overparameterisation” of model parameters, yielding well-supported but erroneous nodes in the tree [23,27–29].
To address these challenges, we developed an extensive statistical pipeline to study the evolutionary patterns in the mitogenomes of Carcharhiniformes (groundsharks), with a particular focus on the contentious Triakidae family (houndsharks; Linck 1790 [30]) [31,32]. Our overarching goal was to evaluate biological signatures in the dataset influencing phylogenetic resolution before selecting the optimal phylogenetic workflow as illustrated in Figure 1. A representative collection of carcharhiniform mitogenomes and outgroups were selected and aligned as described in Section A, whereafter substitution saturation tests were conducted to inform the selection of partitioning schemes as described in Section B. Section C details the mitophylogenomic pipeline used to conduct model selection for ML and Bayesian Inference (BI) analyses and investigate topological conflict around branches of the species tree using concordance factors. Next, the effects of gene-tree conflict on species-tree inference were estimated under the multispecies coalescent model (MSCM) as described in Section D. Lastly, site-heterogeneous models were tested as described in Section E. This protocol can be used to design mitophylogenomic bioinformatic pipelines and serves as educational material for various higher-education modules in molecular evolution.
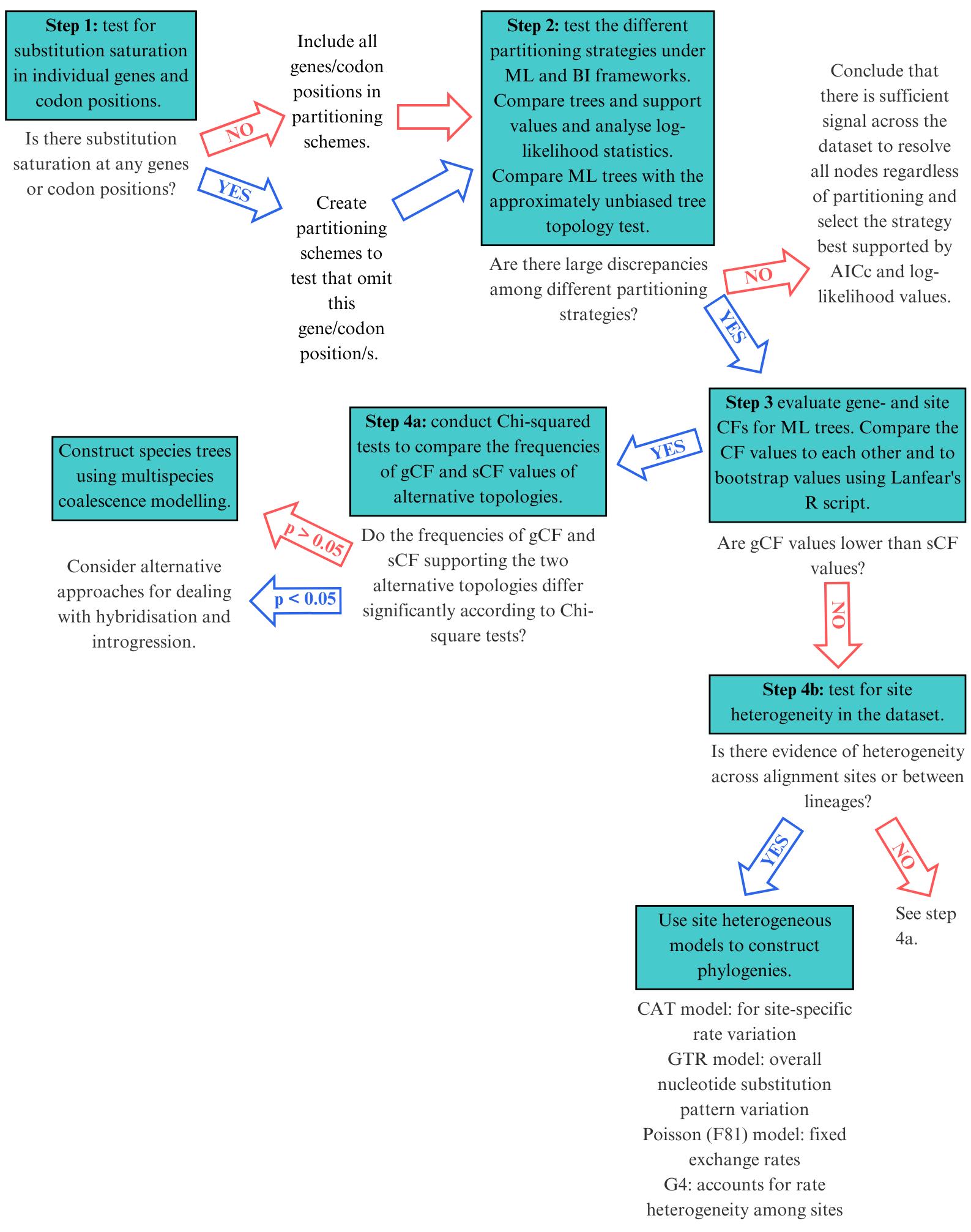
Figure 1. Selecting the right mitophylogenomic approach for your dataset. ML: maximum likelihood; BI: Bayesian inference; gCF: gene concordance factor; sCF: site concordance factor.
Software and datasets
Most of the software programs listed below can be used on Windows 7/8/10/11, Mac OS 10.11 (current versions), and Linux (Ubuntu Desktop LTS, last two supported versions). IQ-Tree has no GUI and must be run through the command line. If you do not have Linux on your device, you can use MobaXTerm v.24.4 (https://mobaxterm.mobatek.net/download.html, last accessed 1/12/2025) for Windows or Tabby Terminal v.1.0.216 (https://tabby.sh/, last accessed 1/12/2025) for Mac to run command line code.
We used a machine with a multi-core central processing unit (CPU) allowing for parallel processing to speed up some of our analyses. The amount of RAM depends on the size of the dataset, but a minimum of 8 GB is recommended. PhyloBayes is primarily developed and distributed for Linux environments. PHYLOBAYES_MPI, as utilised in this protocol, was run through a multicore high-performance computer. The program is designed to take advantage of multiple processors for parallel computing. A multi-core processor or a cluster of machines with MPI (Message Passing Interface) support is recommended for efficient parallelization. The more cores or nodes you can allocate, the faster the analysis may proceed.
If high-performance computing (HPC) resources are not available, the CIPRES (Cyberinfrastructure for Phylogenetic Research) Science Gateway portal v.3.3 at the San Diego Supercomputer Centre [33] (https://www.phylo.org/, last accessed 1/12/2025) is an online platform that provides a user-friendly web interface for performing computationally intensive phylogenetic analyses. MrBayes, PhyloBayes, MAFFT, IQ-Tree, and more can be run through CIPRES. There is also a Geneious plugin. Users can subscribe to a three-month free trial with 1,000 CPU hours; thereafter, CPU hours can be purchased.
A. Creating alignment datasets
1. Batch Entrez webserver (https://www.ncbi.nlm.nih.gov/sites/batchentrez, last accessed 1/12/2025) for retrieving GenBank sequences from NCBI
2. GBSEQEXTRACTOR v.0.04 [34] (https://github.com/linzhi2013/gbseqextractor, last accessed 1/12/2025) for extracting genes from GenBank files for alignment. It is run through Biopython [35] (http://www.biopython.org/, last accessed 1/12/2025), which must be pre-installed on your system as per the instructions on the Biopython website. Thereafter, GBSEQEXTRACTOR can be downloaded and saved in the folder where you want to run your analyses
3. MACSE v.2.07 [36] (https://www.agap-ge2pop.org/macsee-pipelines/, last accessed 1/12/2025) for alignment of protein-coding genes. Requires a suitable Java runtime environment
4. MAFFT v.7.299 [37,38] webserver (https://mafft.cbrc.jp/alignment/server/, last accessed 1/12/2025) for alignment of ribosomal RNAs
5. Geneious Prime v.2023.2 [39] (https://www.geneious.com/download/, last accessed 1/12/2025) for alignment editing and comparison. Note that a paid license is required to edit alignments. Mega11 [40] (https://www.megasoftware.net/) is an alternative software that can be used for sequence visualisation and editing. Geneious also provides licenses for students doing courses (see https://www.geneious.com/free-course-license, last accessed 1/12/2025)
6. BMGE v.1.12_1 [41] can be accessed through the NGPhylogeny.fr webserver (https://ngphylogeny.fr/tools/tool/273/form, last accessed 1/12/2025) for cleaning of alignments
7. GenBank files and accession number lists for our dataset are in the folder 2_Galeomorphii_mitogenome_sequences: Data_13_Winn2023_Galeomorphii_mitogenome_seqs, and concatenated datasets can be found in the folder 3_Multiple_sequence_alignments: Data 14–23 on our Dryad Digital Repository (doi: 10.5061/dryad.sj3tx969h, last accessed 1/12/2025) and GitHub (https://github.com/JessWinn/Houndshark-Mitogenomics, last accessed 1/12/2025)
B. Testing for substitution saturation and partitioning the data
1. DAMBE version 7.0.35 [42,43] (http://dambe.bio.uottawa.ca/DAMBE/dambe.aspx, last accessed 1/12/2025) for testing for substitution saturation at different codon positions in different mitogenome regions
2. Multiple sequence alignment datasets for nucleotide and amino acids in fasta format can be found in the folder 3_Multiple_sequence_alignments: Data_14_Galeomorphii_13PCGs_NT.fasta, Data_17_Galeomorphii_13PCGs_2rRNAs_NT.fasta, Data_20_Galeomorphii_13PCGs_AA.fasta on the Dryad Digital Repository (doi: 10.5061/dryad.sj3tx969h) and GitHub (https://github.com/JessWinn/Houndshark-Mitogenomics)
C. Mitophylogenomic reconstruction
1. ModelFinder v.1.6.12 [44] is available through IQ-Tree v.2.1.3 [45] (http://www.iqtree.org/, last accessed 1/12/2025) for data partitioning and evolutionary model selection and maximum likelihood tree construction (with UFBoot2, SH-aLRT, and CF values)
2. Cyberinfrastructure for Phylogenetic Research (CIPRES) Science Gateway portal v.3.3 at the San Diego Supercomputer Centre [33] (https://www.phylo.org/) to run MrBayes v.3.2.6 [46], for Bayesian inference analyses. You may also select to run the program using a multicore high-performance computer if you have access to one
3. R (https://cran.r-project.org/, last accessed 1/12/2025) and R Studio (https://posit.co/products/open-source/rstudio/) for comparing ML tree support values and constructing BI Tracer plots
4. FigTree v.1.4.4 [47] (https://github.com/rambaut/figtree/releases, last accessed 1/12/2025) for tree visualisation. Requires a Java runtime environment
5. Evolview v3 [48] webpage (https://www.evolgenius.info/evolview/, last accessed 1/12/2025) for tree annotation
6. Multiple sequence alignment nucleotide and amino acid datasets in fasta and nexus format can be found in the folder 3_Multiple_sequence_alignments: Data_14_Galeomorphii_13PCGs_NT.fasta, Data_16_Galeomorphii_13PCGs_NT.nex, Data_17_Galeomorphii_13PCGs_2rRNAs_NT.fasta, Data_19_Galeomorphii_13PCGs_2rRNAs_NT.nex, Data_20_Galeomorphii_13PCGs_AA.fasta, and Data_22_Galeomorphii_13PCGs_AA.nex; partition files in nexus format can be found in the folder 4_Partition_files on the Dryad Digital Repository and GitHub
D. Multispecies coalescent modelling
1. IQ-Tree v.2.1.3 [45] (http://www.iqtree.org/, last accessed 1/12/2025) for gene tree construction
2. ASTRAL v.5.6.3 [49] (https://github.com/smirarab/ASTRAL, last accessed 1/12/2025), a summary-based method to estimate the effects of gene-tree conflict on species-tree inference under the multispecies coalescent model. Java 1.6 or later is required
3. Newick Utilities (https://github.com/tjunier/newick_utils, last accessed 1/12/2025) for collapsing branches with low support for input into ASTRAL. See https://github.com/tjunier/newick_utils/blob/master/doc/nwutils_tutorial.pdf (last accessed 1/12/2025) for specific compiler requirements
4. SVDQuartets [50] in PAUP* v4.0a 169 [51] (https://phylosolutions.com/paup-test/, last accessed 1/12/2025), a site-based method to estimate the effects of gene-tree conflict on species-tree inference under the multispecies coalescent model
5. FigTree v.1.4.4 [47] (https://github.com/rambaut/figtree/releases, last accessed 1/12/2025) for tree visualisation. Requires a Java runtime environment
6. Cleaned and edited gene alignments necessary for ASTRAL were generated in Section C and saved in the folder 2c_CleanEdit. A sample nexus file with gene partitions for our dataset needed to run SVDQuartets can be found in the folder 3_Multiple_sequence_alignments: Data_23_13PCGs_2rRNAs_NT_svd_partitions on the Dryad Digital Repository and GitHub
E. Testing site heterogenous models
1. AliGROOVE v.1.08 [2] (https://github.com/PatrickKueck/AliGROOVE, last accessed 1/12/2025) for compositional heterogeneity among lineages and across sites. AliGROOVE is implemented in Perl and uses the Phylo module of the BioPerl library, which is delivered within the package. AliGROOVE GUI is based on C++ and the Qt library
2. BaCoCa v.1.1 [52] (https://github.com/PatrickKueck/BaCoCa, last accessed 1/12/2025) for compositional heterogeneity among lineages and across sites. To execute BaCoCa, a PERL interpreter must be installed. Linux and Mac systems normally contain this as a standard tool, but the additional PERL Statistics::R package must be installed to use the -r option of BaCoCa.vX.X.r.pl, which allows the generation of result heat maps by using R. Windows users have to install a PERL interpreter ex post. The developers recommend ActivePerl (http://activeperl.softonic.de/, last accessed 1/12/2025). The additional PERL package Statistics::R can be installed via the ActivePerl package manager and R can be installed from: http://cran.r-project.org/bin/windows/base/ (last accessed 1/12/2025). See https://github.com/PatrickKueck/BaCoCa/blob/master/BaCoCa_Manual.pdf (last accessed 1/12/2025) for more detailed instructions
3. PHYLOBAYES_MPI v.1.9 package [53] (https://github.com/bayesiancook/pbmpi, last accessed 1/12/2025) for Bayesian inference analyses using the pb_mpi program and various site-heterogeneous models.
4. Tracer v.1.7.1 [54] (https://github.com/beast-dev/tracer/releases/tag/v1.7.1, last accessed 1/12/2025) for visualisation and diagnostics of Markov chain Monte Carlo (MCMC) output requiring Java v.1.6 or greater
5. FigTree v.1.4.4 [47] (https://github.com/rambaut/figtree/releases, last accessed 1/12/2025) for tree visualisation. Requires a Java runtime environment
6. The three alignment datasets in fasta and phylip format are found in the folder 3_Multiple_sequence_alignments: Data_14_Galeomorphii_13PCGs_NT.fasta, Data_15_Galeomorphii_13PCGs_NT.phy, Data_17_Galeomorphii_13PCGs_2rRNAs_NT.fasta, Data_18_Galeomorphii_13PCGs_2rRNAs_NT.phy, Data_20_Galeomorphii_13PCGs_AA.fasta, Data_21_Galeomorphii_13PCGs_AA.phy and the partition file for BaCoCa is found in the folder 4_Partition_files: 13PCGs_2rRNAs_NT.part.txt on the Dryad Digital Repository and GitHub
Procedure
文章信息
稿件历史记录
提交日期: Oct 30, 2024
接收日期: Jan 26, 2025
在线发布日期: Feb 24, 2025
出版日期: Mar 5, 2025
版权信息
© 2025 The Author(s); This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/).
如何引用
Winn, J. C., Bester-van der Merwe, A. E. and Maduna, S. N. (2025). Annotated Bioinformatic Pipelines for Phylogenomic Placement of Mitochondrial Genomes. Bio-protocol 15(5): e5232. DOI: 10.21769/BioProtoc.5232.
分类
生物信息学与计算生物学
系统生物学 > 基因组学 > 种系遗传学
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。