

Annotated Bioinformatic Pipelines for Genome Assembly and Annotation of Mitochondrial Genomes
线粒体基因组组装与注释的生物信息学流程详解
发布: 2025年03月05日第15卷第5期 DOI: 10.21769/BioProtoc.5231 浏览次数: 3103
评审: Waldir Miron Berbel-FilhoPrashanth N SuravajhalaMaria Lynn Spletter
参见作者原研究论文
The authors used this protocol in:

Jan 2024
Advertisement








Abstract
Mitochondrial genomes (mitogenomes) display relatively rapid mutation rates, low sequence recombination, high copy numbers, and maternal inheritance patterns, rendering them valuable blueprints for mapping lineages, uncovering historical migration patterns, understanding intraspecific population dynamics, and investigating how environmental pressures shape traits underpinned by genetic variation. Here, we present the bioinformatic pipeline and code used to assemble and annotate the complete mitogenomes of five houndsharks (Chondrichthyes: Triakidae) and compare them to the mitogenomes of other closely related species. We demonstrate the value of a combined assembly approach for detecting deviations in mitogenome structure and describe how to select an assembly approach that best suits the sequencing data. The datasets required to run our analyses are available on the GitHub and Dryad repositories.
Key features
• Tips and code for conducting de novo, reference-based, and hybrid assembly.
• Guide to detecting deviations in the structure of the mitochondrial genome.
• Step-by-step guide to annotating and comparing the characteristics of mitochondrial genomes.
• Access to the scripts, data files, and pipelines used to enable replication of all analyses.
Keywords: Comparative mitogenomics (比较线粒体基因组学)Graphical overview
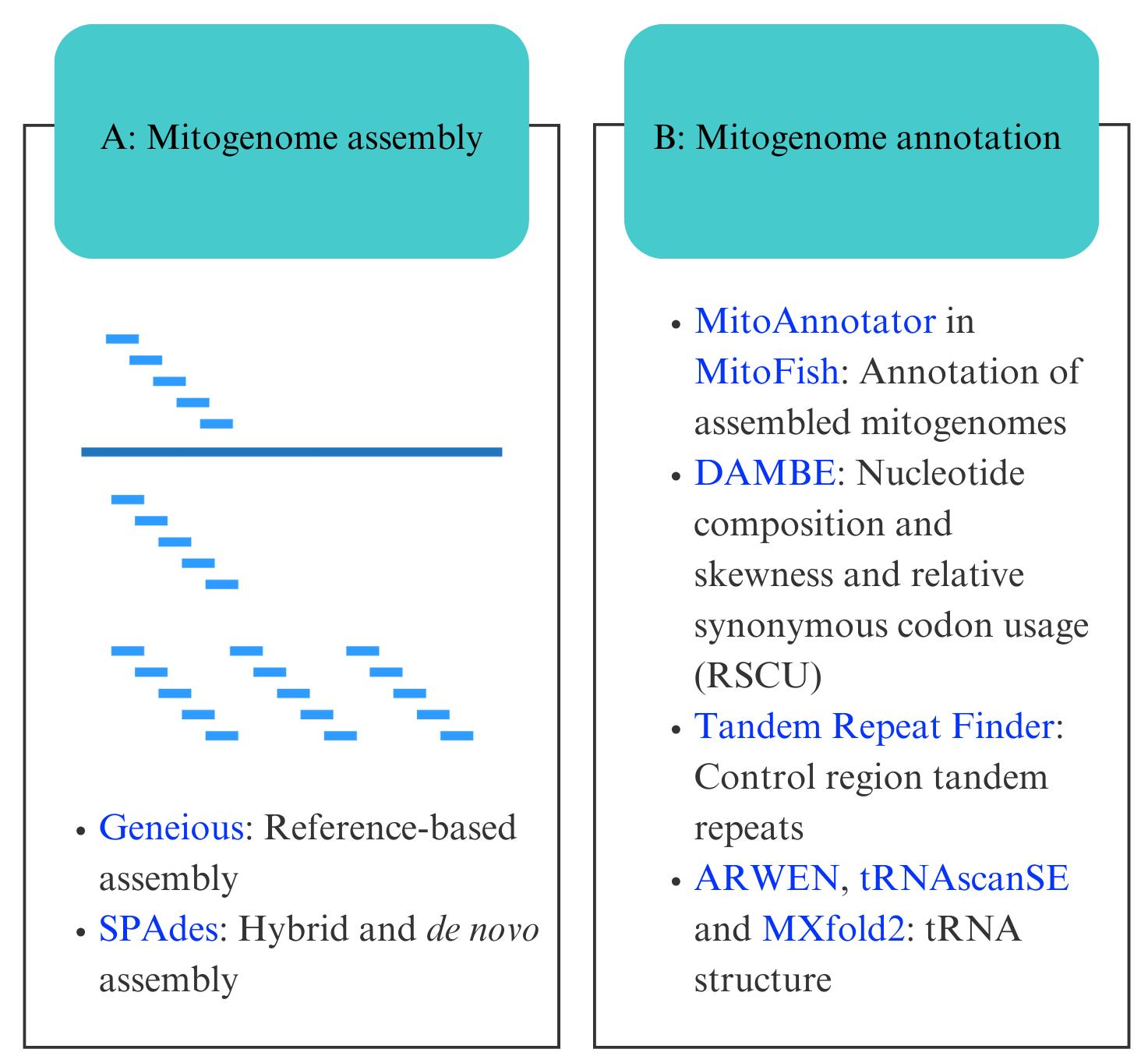
Bioinformatic workflow for mitogenome assembly and annotation. Software programs are indicated in blue.
Background
Mitochondrial genomes (mitogenomes) are found in high copy numbers in the majority of eukaryotic cells and are among the most abundant genome sequences deposited in nucleotide databases [1,2]. In animals, the mitogenome ranges from 14 to over 20 kilobase pairs (kbp) in size and, although gene order can vary, gene content is highly conserved [3,4]. Mitogenomes contain short intergenic spacers, a limited occurrence of gene duplications and rearrangements, and generally lack introns [4,5]. These features make them ideal candidates for investigating complex evolutionary processes at a resolution that far surpasses that of nuclear genomes [4]. Despite gene content being conserved in most species, gene arrangements and duplications can distinguish specific evolutionary lineages, making them useful for resolving taxonomic relationships, conducting population genomics investigations, and contributing to environmental DNA metabarcoding databases [6–8]. Single mitochondrial genes have commonly been used to assess interspecific divergence and intraspecific diversity; however, their phylogenetic signal is transcended by that of the full mitogenome, making mitogenomic analyses more powerful as well as comparable across a wider range of studies [6,9–11].
Genomic studies are being revolutionised by the increasing availability of next-generation sequencing (NGS) technologies, which have enabled the procurement of mitogenomes rapidly and at a low cost [12,13]. However, there is a shortage of standardised pipelines for the full reconstruction and characterisation of these small molecules [14]. The assembly of mitogenomes from high-throughput sequencing (HTS) data can be achieved by mapping reads to a reference mitogenome or by using de novo assembly. Tools such as the Burrows-Wheeler Alignment tool [15], Bowtie2 [16], Minimap2 [17], BBMap [18], and Geneious Prime [19] (https://www.geneious.com/download, last accessed 1/12/2025) can be used for reference-based assembly. This is the less computationally intensive approach; however, it relies on the use of a closely related, well-annotated reference mitogenome to avoid erroneous reconstruction. It may also collapse duplicated regions and fail to detect structural deviations from the reference, particularly when utilizing short reads generated by Illumina or Ion Torrent platforms [20,21]. The alternative approach is a de novo assembly using tools such as SPAdes [22], MitoZ [23], MITObim [24], MitoHiFi [25], and NOVOPlasty [26], which are useful in the absence of a closely related reference or for a contiguous assembly of complex, repetitive, or structurally variable regions of the mitogenome with short reads [21]. Long-read sequencing technologies, including PacBio HiFi sequencing and Oxford Nanopore Technology (ONT) sequencing, circumvent many of these limitations [27,28]. However, a carefully designed assembly approach can ensure a successful assembly of more complex mitogenomes with short-read sequences from Illumina and Ion Torrent platforms.
In this protocol, we describe a three-step mitogenome assembly approach and provide a guide for detecting structural deviations using sequencing data generated for five species belonging to the Triakidae family (houndsharks; Linck 1790 [29]) in Section A. Section B provides a detailed annotation and comparative mitogenomics pipeline. This protocol can be used to design mitogenomic assembly pipelines and serves as educational material for various higher-education modules in molecular evolution.
Software and datasets
Most of the software programs listed below can be used on Windows 7/8/10/11, Mac OS 10.11 (current versions), and Linux (Ubuntu Desktop LTS, last two supported versions). A 64-bit Linux system or Mac OS (with Python 2.7 and Python 3: 3.2 and higher to be pre-installed on it) is required to run SPAdes and Quast. If you do not have Linux on your device, you can use MobaXTerm v.24.4 (https://mobaxterm.mobatek.net/download.html, last accessed 1/12/2025) for Windows or Tabby Terminal v.1.0.216 (https://tabby.sh/, last accessed 1/12/2025) for Mac to run a command line code.
We used a machine with a multi-core central processing unit (CPU) allowing for parallel processing to speed up some of our analyses. The amount of RAM depends on the size of the dataset, but at least 8 GB is recommended. If high-performance computing (HPC) resources are not available, the CIPRES (Cyberinfrastructure for Phylogenetic Research) Science Gateway portal v.3.3 at the San Diego Supercomputer Centre [30] (https://www.phylo.org/, last accessed 1/12/2025) is an online platform that provides a user-friendly web interface for performing computationally intensive phylogenetic analyses. SPAdes can be run through CIPRES, and there is a Geneious plugin too. Users can subscribe to a three-month free trial with 1,000 CPU hours; thereafter, CPU hours can be purchased.
A. Mitogenome assembly
1. FastQC version (v).12.0 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/, last accessed 1/12/2025) for quality control of sequencing files. It requires a suitable Java runtime environment (https://adoptium.net/, last accessed 1/12/2025)
2. Geneious Prime v.2023.2 [19] (https://www.geneious.com/download/, last accessed 1/12/2025) for reference-based assembly and assembly editing and comparison. Note that although reference assembly using the Geneious assembly tool is available in the free version, a paid license is required to edit assemblies or alignments. Mega11 [31] (https://www.megasoftware.net/, last accessed 1/12/2025) is an alternative software that can be used for sequence visualisation and editing. Geneious also provides licenses for students doing courses (see https://www.geneious.com/free-course-license, last accessed 1/12/2025). Alternatively, there are other programs available for reference-based mitogenome assembly (e.g., GetOrganelle [32] and MITObim [24]). The Burrows-Wheeler Alignment tool [15] can also be used to extract mitogenome reads via alignment to a reference, which can then be assembled in SPAdes. Remember to select the software most suited to your organism under study
3. SPAdes v.3.15 [22] (https://github.com/ablab/spades, last accessed 1/12/2025) for hybrid and de novo assembly. SPAdes can now be run in the latest version of Geneious. When you run the SPAdes assembler for the first time, Geneious provides instructions for installing the necessary Windows or Mac features onto your device
4. Quast v.5.0.2 [33] (https://github.com/ablab/quast/releases, last accessed 1/12/2025) for assembly quality checking
5. FinchTV 1.5 (https://digitalworldbiology.com/FinchTV, last accessed 1/12/2025) for sequence trimming for Sanger sequences
6. Our raw Ion Torrent bam files for mitogenome assembly are available on the SRA database (BioProject repository):
a. Data identification number: PRJNA997468 (BioSample accessions: SAMN36680060, SAMN36680061, SAMN36680062, SAMN36680063, SAMN36680064)
b. Direct URL to data (last accessed 1/12/2025): https://www.ncbi.nlm.nih.gov/bioproject/997468; https://www.ncbi.nlm.nih.gov/biosample/36680060; https://www.ncbi.nlm.nih.gov/biosample/36680061; https://www.ncbi.nlm.nih.gov/biosample/36680062; https://www.ncbi.nlm.nih.gov/biosample/36680063; https://www.ncbi.nlm.nih.gov/biosample/36680064
c. Alternatively, filtered reads for the mitogenome can be found in the folder 1_Raw_Ion Torrent_NGS_data: Data_1_Galeorhinus_galeus_Ion Torrent_Filtered_RawData, Data_2_Mustelus_asterias_Ion Torrent_Filtered_RawData, Data_3_Mustelus_mosis_Ion Torrent_Filtered_RawData, Data_4_Mustelus_palumbes_Ion Torrent_Filtered_RawData, Data_5_Triakis_megalopterus_Ion Torrent_Filtered_RawData on the Dryad Digital Repository (doi: 10.5061/dryad.sj3tx969h, last accessed 1/12/2025)
B. Mitogenome annotation
1. MitoAnnotator in MitoFish v.3.72 webserver [34,35] (http://mitofish.aori.u-tokyo.ac.jp/annotation/input/, last accessed 1/12/2025) for mitogenome annotation
2. Sequence Manipulation Suite 2 [36] (https://www.bioinformatics.org/sms2/translate.html, last accessed 1/12/2025) for checking the reading frame of protein-coding genes
3. GenBank [37] submission platform (https://www.ncbi.nlm.nih.gov/WebSub/, last accessed 1/12/2025) for submission of GenBank files
4. DAMBE v.7.0.35 [38,39] (http://dambe.bio.uottawa.ca/DAMBE/dambe.aspx, last accessed 1/12/2025) for calculating nucleotide composition and skewness and relative synonymous codon usage
5. R (https://cran.r-project.org/, last accessed 1/12/2025) and R Studio (https://posit.co/products/open-source/rstudio/, last accessed 1/12/2025) for constructing nucleotide composition, skewness, and RSCU plots
6. ARWEN v.1.2.3 webserver [40] (http://130.235.244.92/ARWEN/, last accessed 1/12/2025), tRNAscanSE webserver v.2.0 [41] (http://lowelab.ucsc.edu/cgi-bin/tRNAscan-SE2.cgi, last accessed 1/12/2025), and MXfold2 [42] webserver (http://ws.sato-lab.org/mxfold2/, last accessed 1/12/2025) for checking tRNA folding
7. Tandem Repeat Finder [43] (https://tandem.bu.edu/trf/trf.html, last accessed 1/12/2025) for characterising repeats in the control region
8. Mitogenome sequence files for our Triakidae mitogenomes can be found on GenBank. Data identification numbers: ON075075, ON075076, ON075077, ON652873, and ON652874. Direct URL to data (last accessed 1/12/2025): https://www.ncbi.nlm.nih.gov/nuccore/ON075075; https://www.ncbi.nlm.nih.gov/nuccore/ON075076; https://www.ncbi.nlm.nih.gov/nuccore/ON075077; https://www.ncbi.nlm.nih.gov/nuccore/ON652873; https://www.ncbi.nlm.nih.gov/nuccore/ON652874
a. Alternatively, these have been uploaded to the folder 2_Galeomorphii_mitogenome_sequences: Data_8_ON652873_Mustelus_asterias, Data_9_ON652874_Galeorhinus_galeus, Data_10_ON075075_Triakis_megalopterus, Data_11_ON075076_Mustelus_palumbes, Data_12_ON075077_Mustelus_mosis in on our Dryad Digital Repository (doi: 10.5061/dryad.sj3tx969h, last accessed 1/12/2025)
Procedure
文章信息
稿件历史记录
提交日期: Oct 30, 2024
接收日期: Jan 26, 2025
在线发布日期: Feb 24, 2025
出版日期: Mar 5, 2025
版权信息
© 2025 The Author(s); This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/).
如何引用
Winn, J. C., Bester-van der Merwe, A. E. and Maduna, S. N. (2025). Annotated Bioinformatic Pipelines for Genome Assembly and Annotation of Mitochondrial Genomes. Bio-protocol 15(5): e5231. DOI: 10.21769/BioProtoc.5231.
分类
生物信息学与计算生物学
系统生物学 > 基因组学 > 种系遗传学
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。