

Optimal Dual RNA-Seq Mapping for Accurate Pathogen Detection in Complex Eukaryotic Hosts
优化双RNA-Seq比对方法以精准检测复杂真核宿主中的病原体
发布: 2025年02月05日第15卷第3期 DOI: 10.21769/BioProtoc.5182 浏览次数: 2683
评审: Prashanth N SuravajhalaSoumya MoonjelyWilly R Carrasquel-UrsulaezAnonymous reviewer(s)
Advertisement










Abstract
Dual RNA-Seq technology has significantly advanced the study of biological interactions between two organisms by allowing parallel transcriptomic analysis. Existing analysis methods employ various combinations of open-source bioinformatics tools to process dual RNA-Seq data. Upon reviewing these methods, we intend to explore crucial criteria for selecting standard tools and methods, especially focusing on critical steps such as trimming and mapping reads to the reference genome. In order to validate the different combinatorial approaches, we performed benchmarking using top-ranking tools and a publicly available dual RNA-Seq Sequence Read Archive (SRA) dataset. An important observation while evaluating the mapping approach is that when the adapter trimmed reads are first mapped to the pathogen genome, more reads align to the pathogen genome than the unmapped reads derived from the traditional host-first mapping approach. This mapping method prevents the misalignment of pathogen reads to the host genome due to their shorter length. In this way, the pathogenic read information found at lesser proportions in a complex eukaryotic dataset is precisely obtained. This protocol presents a comprehensive comparison of these possible approaches, resulting in a robust unified standard methodology.
Key features
• Benchmarking of top-ranking software for quality control, adapter trimming, and read mapping.
• Emphasizes the importance of read mapping criteria for dual RNA-Seq datasets: (i) high count of uniquely host mapped reads, (ii) low count of host multi-mapped reads, and (iii) high count of unmapped reads belonging to pathogens.
• Elaborates the best mapping approach to precisely extract the pathogen reads as these get captured comparatively less in dual RNA-Seq datasets.
Keywords: Dual RNA-Seq (双RNA-Seq)Graphical overview
Background
Dual RNA sequencing is a powerful tool to precisely investigate the complete gene expression profile of two actively interacting organisms. When a pathogen infects a host, both develop adaptation mechanisms by revealing a series of changes at molecular levels. These biological changes are interrelated and can be effectively elucidated by analyzing the whole transcriptome data of the host–pathogen interaction. Until 2012, transcriptomics sequencing of infectious diseases was limited to separately capturing the cascade of biological events in either the host or the pathogen [1]. This allows us to study gene expression changes only from a linear perspective. Over the years, insightful research in transcriptomics analysis has encouraged researchers across the globe to devise the concept of extracting and sequencing whole RNA from the infection site. This idea will provide a broader scope for unraveling the role of smaller molecules and studying the biological changes in organisms at different stages of interaction. Compared to conventional RNA sequencing technology, dual RNA sequencing caters to a sufficient amount of host and pathogen RNA and other small RNA molecules (microRNA, long non-coding RNA, and other non-coding RNA) when pathogen-infected samples are subjected to the well-established and published methods of RNA extraction and sequencing procedures [2–5].
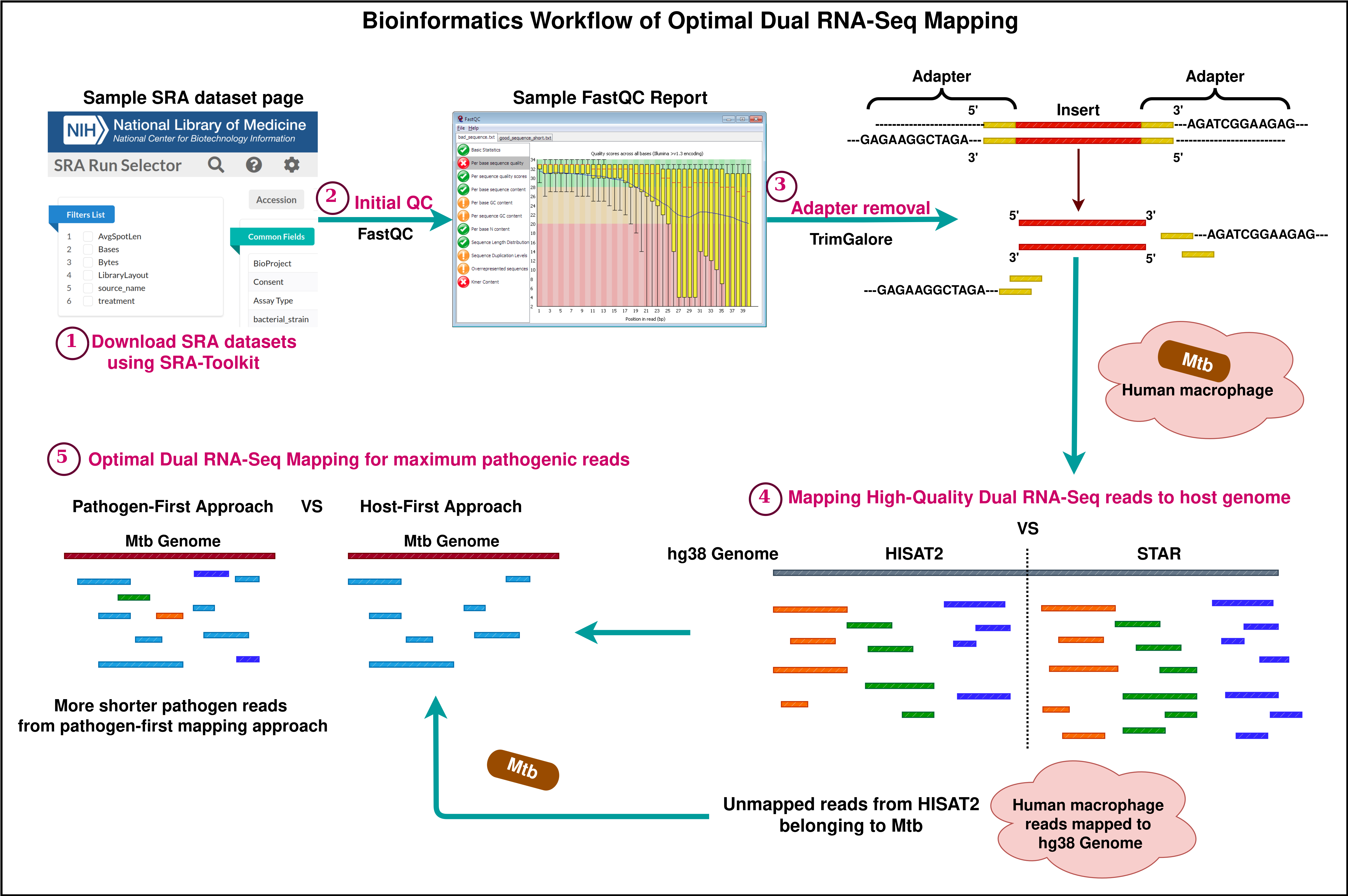
Dual sequencing data generated as reads or fragments comprises information on both the host and the pathogen. The infected host cells may not always reflect pathogen-related data in enormous quantities. Therefore, a detail-oriented analysis is necessary to capture the minimally found essential information on host–pathogen interplay. Several bioinformatics analysis methods and tools are available to explore the complex datasets involving eukaryotic and prokaryotic reads [6–8]. Most of these methods suggest a sequential mapping approach to extract host and pathogen reads. Some studies recommend a combined mapping approach where the reference genomes of the host and pathogen are concatenated, indexed, and used as a single reference [9,10]. As far as quality control and adapter removal are concerned, standard methods for read mapping have been receiving greater attention as pathogen data is found in lesser quantities. Espindula et al. [10] conducted studies on different infection models and showed that other alternative mapping approaches outperform the traditional host-first mapping approach. When trimmed reads are mapped first to the host reference, there are high chances of pathogen read mismapping due to its shorter read length. To avoid this, alternative mapping approaches were introduced, and this bio-protocol emphasizes one such mapping technique—the pathogen-first mapping approach—in detail. A comparison of mapping results proved that most of the pathogen reads have been restored in the pathogen-first mapping approach. The results obtained using this approach are now based on a higher confidence scale and can be used for further processing.
This protocol is presented with the aim of demonstrating a standardized bioinformatics procedure for productive mapping. In this protocol, human monocyte-derived macrophages (HMDMs) infected with Mycobacterium tuberculosis (Mtb) were used as a dual RNA-Seq model. Apart from humans infected with Mtb, other host and pathogen models can still be used in dual RNA-Seq experiments. For example, other dual RNA-Seq datasets are publicly available in Gene Expression Omnibus (GEO); one such dataset is Triticum aestivum infected with Fusarium graminearum (Accession: SRP439529; GEO: GSE233409). Dual RNA-Seq test dataset of human–Mtb was downloaded from Sequence Read Archive (SRA), National Centre for Biotechnology Information (Accession: SRP359986 [11]). This study primarily focuses on a compound that restricts Mycobacterium tuberculosis from catabolizing cholesterol by binding with iron. The data quality control before and after read trimming was performed using FastQC. The FastQC tool can be accessed online at https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. For trimming low-quality bases and adapter removal, benchmarking was performed using the topmost adapter trimming software, namely fastp [12] and Trim-Galore (https://github.com/FelixKrueger/TrimGalore). Following this, the topmost splice-aware alignment tools STAR [13] and HISAT2 [14] were also benchmarked to obtain optimal results. The most important part of the analysis is to apply the best mapping method that serves the experimental purpose of dual RNA-Seq analysis. After mapping the reads to their respective genomes, the reads mapping to genes were quantified using featureCounts [15]. Other downstream analysis methods, which include read count normalization, differential expression analysis, gene ontology, and pathway enrichment, are not demonstrated here, as the key aim of this protocol is to highlight the crucial preparatory steps like quality control, adapter removal, and read mapping in a descriptive manner.
Software and datasets
1. Data
Dual RNA-Seq datasets are publicly available on NCBI Sequence Read Archive (SRA) and can be downloaded and analyzed for learning purposes. For this protocol, a dataset from SRA (accession: SRP359986, Gene Expression Omnibus datasets; GEO: GSE196816) was downloaded and utilized.
2. Bioinformatics tools (all tools were installed using conda)
• SRA-Toolkit (version 3.1.0) includes tools like prefetch and fasterq-dump for fetching SRA datasets and extracting individual fastq files.
• FastQC (version 0.12.1). For assessing the quality of fastq reads before and after trimming.
• MultiQC (version 1.19). For integrating results into interactive visualization reports throughout the analysis.
• TrimGalore (version 0.6.10) and Cutadapt (version 4.6). For quality-trimming bases from reads, automatic adapter detection and removal, and filtering reads based on lengths.
• HISAT2 (version 2.2.1). For indexing the reference genome and mapping trimmed high-quality reads to the reference genome of eukaryotes.
• BWA (version 0.7.17-r1188). For indexing the reference genome and mapping trimmed high-quality reads to the reference genome of prokaryotes.
• SAMtools (version 1.19). For converting huge files from mapping results (.sam) into binary formatted .bam files enabling easy processing, to sort reads with their mate pairs, and to check the statistical distribution of reads after mapping.
• Bedtools (version v2.31.1). For extracting the interleaved reads inside .bam files into paired-end separate fastq files.
• featureCounts from Subread package (version v2.0.6). For quantifying all reads mapped to genomic coordinates using annotation feature file(.gtf/.gff3) of the reference genome.
3. Platform used: Linux, Ubuntu
• CPU: Architecture, 64 bit; 24 cores, 96 threads
• Memory: 512 GB RAM
Note: The threads/cores mentioned in each step of the analysis need to be modified by users as per the computational resources available.
Procedure
文章信息
稿件历史记录
提交日期: Aug 28, 2024
接收日期: Dec 8, 2024
在线发布日期: Dec 26, 2024
出版日期: Feb 5, 2025
版权信息
© 2025 The Author(s); This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/).
如何引用
Eden M., I. S. T. and Vetrivel, U. (2025). Optimal Dual RNA-Seq Mapping for Accurate Pathogen Detection in Complex Eukaryotic Hosts. Bio-protocol 15(3): e5182. DOI: 10.21769/BioProtoc.5182.
分类
生物信息学与计算生物学
系统生物学 > 转录组学 > RNA测序
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。