

Classification of a Massive Number of Viral Genomes and Estimation of Time of Most Recent Common Ancestor (tMRCA) of SARS-CoV-2 Using Phylodynamic Analsysis
使用系统动力学对大量病毒基因组分类并预估SARS-CoV-2最近共同祖先年代(tMRCA)
(*contributed equally to this work) 发布: 2024年03月20日第14卷第6期 DOI: 10.21769/BioProtoc.4955 浏览次数: 2656
评审: Migla MiskinytePooja VermaAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Jun 2023
Advertisement











Abstract
Estimating the time of most recent common ancestor (tMRCA) is important to trace the origin of pathogenic viruses. This analysis is based on the genetic diversity accumulated in a certain time period. There have been thousands of mutant sites occurring in the genomes of SARS-CoV-2 since the COVID-19 pandemic started; six highly linked mutation sites occurred early before the start of the pandemic and can be used to classify the genomes into three main haplotypes. Tracing the origin of those three haplotypes may help to understand the origin of SARS-CoV-2. In this article, we present a complete protocol for the classification of SARS-CoV-2 genomes and calculating tMRCA using Bayesian phylodynamic method. This protocol may also be used in the analysis of other viral genomes.
Key features
• Filtering and alignment of a massive number of viral genomes using custom scripts and ViralMSA.
• Classification of genomes based on highly linked sites using custom scripts.
• Phylodynamic analysis of viral genomes using Bayesian evolutionary analysis sampling trees (BEAST).
• Visualization of posterior distribution of tMRCA using Tracer.v1.7.2.
• Optimized for the SARS-CoV-2.
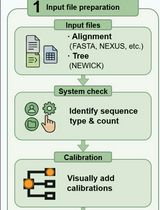
Graphical overview
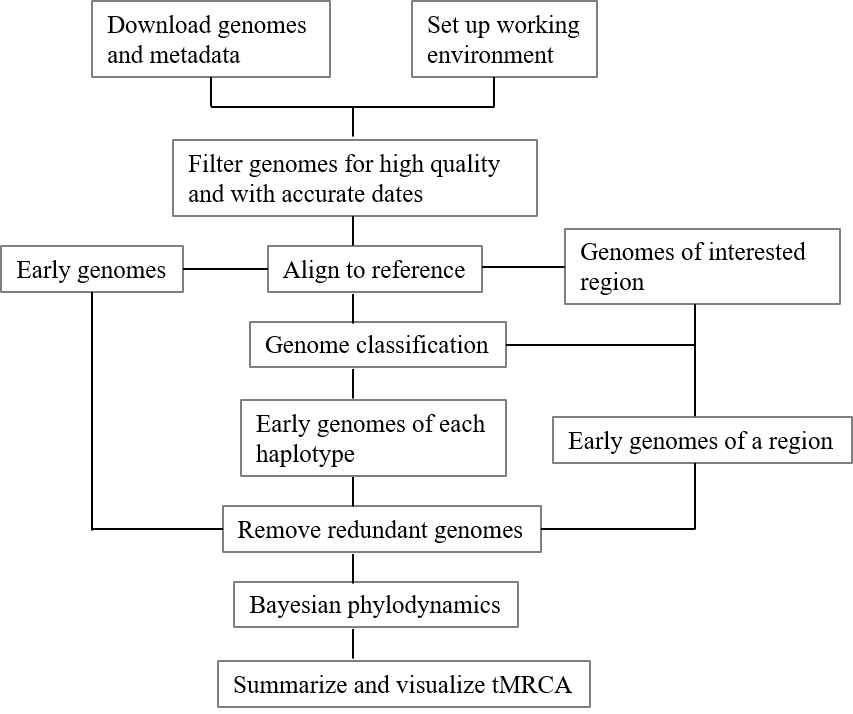
Graphical workflow of time of most recent common ancestor (tMRCA) estimation process
Background
Revealing the origins of pathogenic viruses, crucial for cutting them off from the root and preventing future spillover, requires long-term hard work from scientists all around the world [1]. Although some infectious pathogens can be traced back decades, the debate on their origin continues. For example, AIDS was officially reported on June 5, 1981, by the Centers for Disease Control and Prevention of the USA. Five years later, HIV infection was detected in a human serum sample collected in Léopoldville in early 1959 [2]. Bayesian phylodynamic analyses using recovered viral gene sequences from decades-old paraffin-embedded tissues traced the most recent common ancestor (MRCA) of the M group of HIV back to approximately 1908 (CI 1884–1924), suggesting that HIV has been circulating in the human population for approximately 100 years [3]. MERS-CoV is another example, as it was first reported in a Saudi Arabian man in 2012 [4]. Bats are thought to be the reservoir hosts of MERS-CoV, and dromedary camels are considered to be the major intermediate host [5]; however, the transmission route from animals to humans is not well understood. Researchers tested 189 camel serum samples from 1983 to 1997 and found that 81% had neutralizing antibodies against MERS-CoV, suggesting long-term virus circulation in these animals [6]. Similarly, COVID-19 was first reported on December 27, 2019, in Wuhan, China [7,8], and the Huanan seafood market was suspected to be the place of origin [9]; however, disputes remain. Pekar and colleagues explored the evolutionary dynamics of the first wave of SARS-CoV-2 infections in China using a strict clock Bayesian phylodynamic analysis but failed to capture the index case [10], probably because the redundant sequences were not removed, which usually influences the accuracy of time of MRCA (tMRCA) estimation, as indicated in two recent tMRCA analysis [11,12]. Genome classification plays a critical role in tracing the origin of pathogenic viruses [3,12]. We have previously classified SARS-CoV-2 genomes based on two amino acids, Spike-614 and Orf8-84, and revealed 16 haplotypes. From those, three major haplotypes were found to separately drive the development of the pandemic in China and the world. However, genome classification based on amino acid mutations did not rule out recombination and reverse mutations. In this paper, we provide detailed protocols to filter and classify the massive number of viral genomes according to six highly linked mutations that happened in the early phase of the epidemic by custom scripts and common programs and estimate tMRCA of non-redundant genome subpools.
Materials and reagents
SARS-CoV-2 genome sequences were obtained from the GISAID database [13] on May 1, 2022. Genomes that were collected from hosts other than humans and/or had a length of less than 2,900 nucleotides and more than 0.05% of unknown nucleotides were filtered out. More than 5 million genomes were retained for further analysis.
Equipment
Bioinformatic platform (CentOS, 7.2.1511)
Windows desktop computers (v11, 6/6/2021)
Software and datasets
Perl (v5.34.0, 20/5/2021)
Python (v3.9.0, 5/10/2020)
Gcc (v8.4.1, 28/9/2020)
SeqKit (v2.3.0, 12/8/2022)
minimap2 (v2.24, 26/12/2021)
ViralMSA (v3, 29/6/2007)
cd-hit (v4.8.1, 1/9/2018)
ALTER (v1.3.4, 30/10/2016)
BEAST (v1.10.4, 9/11/2018)
Jdk (v17_windows-x64_bin, 16/6/2021)
Jre (8u341-windows-x64, 5/6/2021)
Tracer (v1.7.2, 5/1/2018)
Global Initiative of Sharing All Influenza Data (GISAID) (https://gisaid.org/) (Access date, 1/5/2022)
All personalized scripts have been deposited in GitHub: https://github.com/XiaowenH/SARS-CoV-2-Classification (Access date, 5/9/2023). Figure 1 is a screenshot of the files in GitHub.
Figure 1. Screenshot of the personalized scripts deposited in GitHub
Procedure
文章信息
版权信息
© 2024 The Author(s); This is an open access article under the CC BY license (https://creativecommons.org/licenses/by/4.0/).
如何引用
Hu, X., Guan, S., He, Y., Yi, G., Yao, L. and Zhang, J. (2024). Classification of a Massive Number of Viral Genomes and Estimation of Time of Most Recent Common Ancestor (tMRCA) of SARS-CoV-2 Using Phylodynamic Analsysis. Bio-protocol 14(6): e4955. DOI: 10.21769/BioProtoc.4955.
分类
生物信息学与计算生物学
系统生物学 > 基因组学 > 种系遗传学
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。