

CoCoNat: A Deep Learning–Based Tool for the Prediction of Coiled-coil Domains in Protein Sequences
CoCoNat:一种基于深度学习的工具,用于预测蛋白质序列中的卷曲螺旋结构域
发布: 2024年02月20日第14卷第4期 DOI: 10.21769/BioProtoc.4935 浏览次数: 2579
评审: Prashanth N SuravajhalaAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Aug 2023
Advertisement










Abstract
Coiled-coil domains (CCDs) are structural motifs observed in proteins in all organisms that perform several crucial functions. The computational identification of CCD segments over a protein sequence is of great importance for its functional characterization. This task can essentially be divided into three separate steps: the detection of segment boundaries, the annotation of the heptad repeat pattern along the segment, and the classification of its oligomerization state. Several methods have been proposed over the years addressing one or more of these predictive steps. In this protocol, we illustrate how to make use of CoCoNat, a novel approach based on protein language models, to characterize CCDs. CoCoNat is, at its release (August 2023), the state of the art for CCD detection. The web server allows users to submit input protein sequences and visualize the predicted domains after a few minutes. Optionally, precomputed segments can be provided to the model, which will predict the oligomerization state for each of them. CoCoNat can be easily integrated into biological pipelines by downloading the standalone version, which provides a single executable script to produce the output.
Key features
• Web server for the prediction of coiled-coil segments from a protein sequence.
• Three different predictions from a single tool (segment position, heptad repeat annotation, oligomerization state).
• Possibility to visualize the results online or to download the predictions in different formats for further processing.
• Easy integration in automated pipelines with the local version of the tool.
Graphical overview
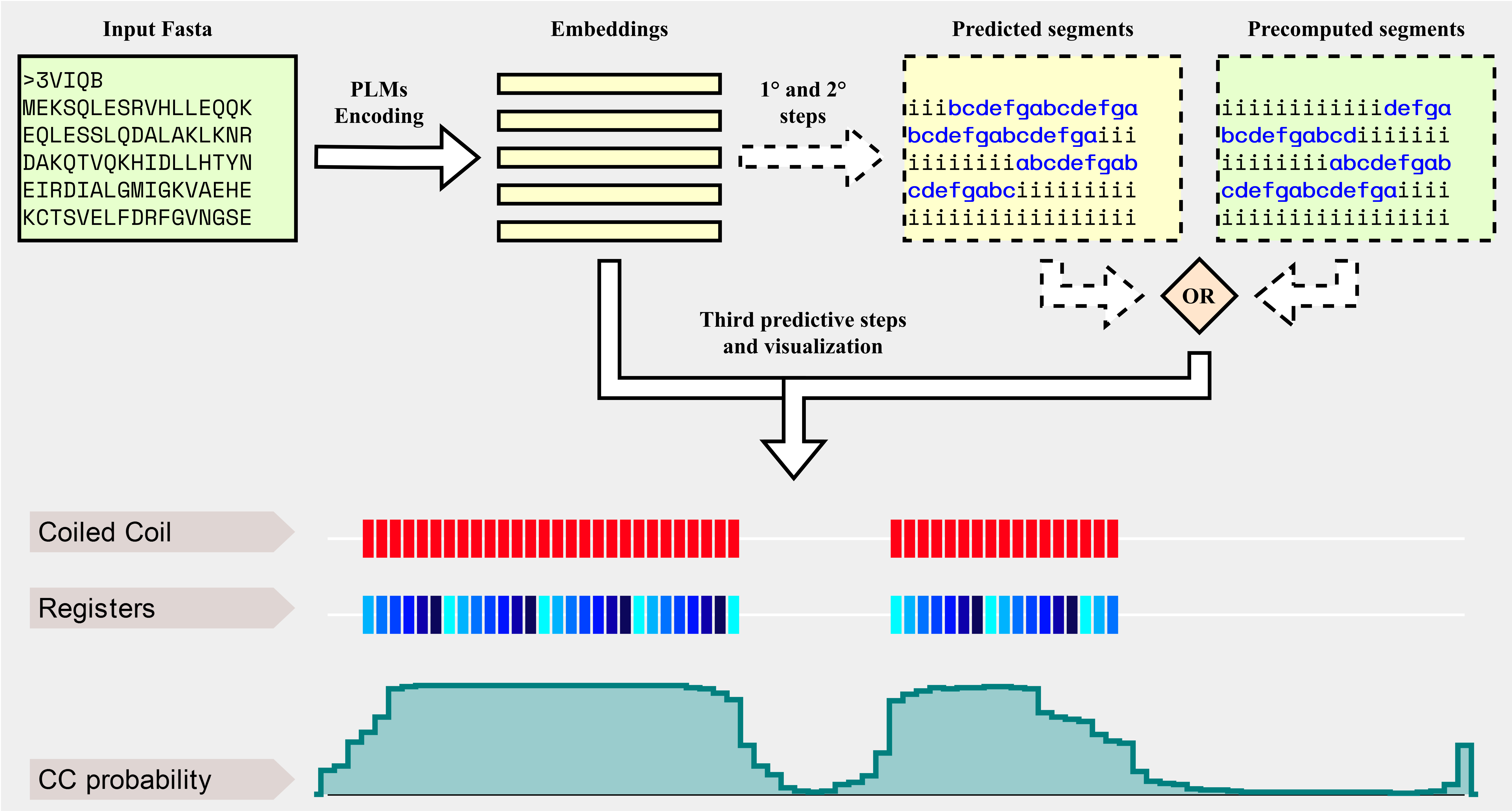
Background
Coiled-coil domains (CCDs) are structural motifs where α‐helices pack together in an arrangement called knobs-into-holes [1], by which residues from one helix (the knobs) pack into holes formed by side chains in the other helices participating in the domain. CCDs have been observed in different kinds of proteins sequenced from all the kingdoms of life [2] and perform a great number of diverse functions.
Canonical CCDs include the interaction of two or more α‐helices, each characterized by the repetition of a seven-residue motif called heptad repeat. The positions of the heptad repeat are referred to as registers and are labeled with the letters a–g. CCDs can be classified into different oligomerization states, depending on the number (dimers, trimers, tetramers, and higher orders) and orientation (parallel or antiparallel) of the involved α‐helices.
Methods such as SOCKET [3] or SamCC-Turbo [4] can annotate CCDs starting from the experimental 3D structure of a protein. In the absence of structural information, several tools have been proposed over the years to perform automatic annotations on protein sequences, each addressing different tasks of CCD prediction (i.e., segment localization, heptad repeat annotation, oligomerization state classification).
Recently, the development of protein language models (PLMs) introduced a novel way of generating embeddings to encode protein sequences for downstream predictive tasks. We proposed CoCoNat [5], a deep learning–based approach that exploits two different and complementary PLMs, ProtT5 [6] and ESM2 [7], to produce a predictive pipeline for the complete ab initio annotation of CCDs.
CoCoNat processes input sequences with three cascading networks, each trained independently to solve a specific task. The first step adopts a deep architecture based on convolutional and recurrent layers to identify the presence of coil-coiled segments along the sequence. The second step adopts a probabilistic graphical model to assign registers to each residue in the segment. Finally, the third step adopts a neural network to predict the oligomerization state of each segment. Each prediction is also complemented with the probabilities computed by the network, allowing users to assess their reliability.
CoCoNat is trained on a dataset comprising 2,198 proteins annotated with 4,342 helices and 9,062 proteins without CCD. When tested on a non-redundant benchmark dataset, comprising 400 proteins annotated with 863 helices and 318 proteins without CCD, CoCoNat outperforms other methods on all three predictive tasks included in the pipeline. Specifically, it achieves a 0.54 per-residue F1 score and a 0.49 per-segment F1 score on the identification of segment boundaries over the sequence (first step in the Graphical overview), a Matthew's Correlation Coefficient (MCC) between 0.83 and 0.84 for each type of register in the annotation of the heptad repeats (second step in the Graphical overview), and an average MCC of 0.58 for the 4-class classification of the oligomerization state (third step in the Graphical overview).
Moreover, the adoption of PLMs to encode the input allows CoCoNat to be extremely time efficient. When tested on the virtual machine hosting the web server (AMD EPYC 7301 12-Core Processor, 48 GB RAM, no GPU), CoCoNat requires an average running time of 330 s (5.5 min) to predict 100 sequences of length comprised between 100 and 200 residues. The same computation takes approximately 2.5 h with CoCoPRED [8], a similar tool based on canonical multiple sequence alignments.
Here, we illustrate in detail how CoCoNat can be adopted as a web server or as a standalone tool, allowing for easy integration into any computational pipeline. As a test case, we select one of the proteins belonging to our benchmark dataset, the Mating-type switching protein swi5 from the organism Schizosaccharomyces pombe (UniProt accession: Q9UUB7). This protein presents two coiled-coil segments organized as a parallel dimer that CoCoNat identifies. Both the registers and the oligomerization classes are correctly assigned. The only difference between the putative and the real annotations is the length of the segments.
Supplementary File S1 reports a schema of the web server compliant with the Minimum Information About Bioinformatics investigation (MIABi) guidelines [9].
Equipment
Computer with internet access and a web browser
(Only for online execution) CoCoNat web server (https://coconat.biocomp.unibo.it/)
(Only for local execution) Machine with a macOS or Linux operating system and at least 4 CPU cores and 48 GB of RAM
The local release is not suitable to be executed directly on a machine with a Windows operating system. In this case, the adoption of a virtual machine or the Windows Subsystem for Linux (WSL) is recommended.
Software and datasets
Docker Engine, installed (https://docs.docker.com/engine/install/debian/)
Miniconda, installed (https://docs.conda.io/projects/miniconda/en/latest/miniconda-install.html)
As long as Miniconda is installed, Python 3 and pip (both mentioned in the protocol) do not need to be installed separately, as they will be included in the environment generated by Miniconda.
Procedure
文章信息
版权信息
© 2024 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
如何引用
Manfredi, M., Savojardo, C., Martelli, P. L. and Casadio, R. (2024). CoCoNat: A Deep Learning–Based Tool for the Prediction of Coiled-coil Domains in Protein Sequences. Bio-protocol 14(4): e4935. DOI: 10.21769/BioProtoc.4935.
分类
生物信息学与计算生物学
生物物理学 > 大分子模拟
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。