

A Post-translational Modification–enhanced Pull-down Method to Study Degron Domains and the Associated Protein Degradation Complexes
一种翻译后修饰增强的牵引法,用于研究 Degron 结构域和相关的蛋白质降解复合物
发布: 2023年09月20日第13卷第18期 DOI: 10.21769/BioProtoc.4816 浏览次数: 2509
评审: Ralph Thomas BoettcherQingliang ShenAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Apr 2023
Advertisement









Abstract
The identification and characterization of the ubiquitin E-ligase complexes involved in specific proteins’ degradation via the ubiquitin-proteasome system (UPS) can be challenging and require biochemical purification processes and in vitro reconstitution assays. Likewise, evaluating the effect of parallel phosphorylation and ubiquitination events occurring in vivo at dual phospho/ubiquitin-regulated motifs (called Phospho-Degrons or pDegrons) driving UPS degradation of the targeted protein has remained elusive. Indeed, the functional study of such E1-E2-E3 complexes acting on a protein-specific level requires previously or otherwise acquired knowledge of the nature of such degradation complex components. Furthermore, the molecular basis of the interaction between an E3 ligase and its pDegron binding motif on a target protein would require individually optimized in vitro kinase and ubiquitination assays. Here, we describe a novel enzymatically enhanced pull-down method to functionally streamline the discovery and functional validation of the ubiquitin E-ligase components interacting with a phospho-degron containing protein domain and/or sub-domain. The protocol combines key features of a protein kinase and ubiquitination in vitro assay by including them in a pull-down step exerted by a known or putative pDegron-tagged peptide using the cell extracts as a source of enzymatically active post-translational modification (PTM) modifying/binding native proteins. The same method allows studying specific stimuli or treatments towards the recruitment of the molecular degradation complex at the target protein’s phospho-degron site, reflecting in vivo–initiated events further enhanced through the assay design. In order to take full advantage of the method over traditional protein–protein interaction methods, we propose to use this PTM-enhanced (PTMe) pull down both towards the degradation complex discovery/ID phase as well as for the functional pDegron recruitment validation phase, which is the one described in the present protocol both graphically and in a stepwise fashion for reproduceable results.
Key features
• Suitable to study UPS-regulated (a) cytosolic and/or nuclear proteins, (b) intracellular region of transmembrane proteins, and (c) protein sub-domains bearing a known/putative pDegron motif.
• Requires a biotin-tagged recombinant version of the target protein and/or sub-domain.
• Allows the qualitative and quantitative analysis of endogenous ubiquitin (Ub) E-ligases recruitment to a known or putative pDegron bearing protein/sub-domain.
• Allows simultaneous testing of various treatments and/or conditions affecting the phosphorylative and/or ubiquitylation status of the studied pDegron bearing protein/sub-domain and the recruited factors.
Graphical overview

Background
Regulation of protein degradation is particularly relevant for the cell to control the expression levels of regulatory proteins along with the underlying biological processes. The major known cellular proteostasis mechanism is mediated by the ubiquitin-proteasome system (UPS) and uses a dedicated set of widespread enzymes called ubiquitin E-ligases. Their role is to flag specific proteins by adding a ubiquitin moiety to specific lysine residues that will then be further identified, targeted, and complexed by E-ligases (typically initiated by E3, followed by E2 and E1 types) and ultimately degraded by the UPS (reviewed in [1]). The essential amino-acid sequence allowing ubiquitin-targeted protein degradation has been called Degrons [2–4]. Degrons in which phosphorylation residue(s) play either a permissive or inhibitory role towards degradation are best defined as phospho-(regulated) Degrons and are ubiquitously found in regulatory proteins involved in growth and developmental processes [4,5]. Phospho-Degrons in which the alternative phosphorylation vs. ubiquitylation requirement establishes a biological switch are also referred as phospho-inhibited degrons [6]. Phospho-Degron motifs can be predicted by the parallel presence of in vivo PTM-modified Ser/Thr/Tyr amino-acid residues within a relative short distance from an in vivo modified lysine [7–9]. At present, the post-translational modification (PTM) status of putative Phospho-Degrons can be exerted by independent assays looking either at the ubiquitylation (in vitro ubiquitination assay) or phosphorylation status (through in vitro kinase assays). We have designed an integrated strategy and method to enhance specificity towards both (a) identifying a putative phospho-degron and simultaneously (b) functionally confirm the ID of the ubiquitin E1,2,3-ligases recruited by the phospho-degron-bearing protein/domain. Central to the described method is the synthesis of a biotin-tagged protein domain/subdomain or peptide bearing the native ubiquitin-binding motif amino acidic sequence for the putative phospho-Degron. This recombinant biotin-tagged protein domain/peptide is used as the bait component of the PTM-enhanced (PTMe) pull-down method. The method relies on the ex vivo–enhanced phosphorylation and ubiquitylation (PTMs) events induced in vivo. This bears increased advantages over performing separate co-immunoprecipitations (co-IP)/far westerns and individual kinase or ubiquitylation assays in immunoprecipitated proteins, as shown in Figure 1. The method can indeed be used prior to further refining individual protein interactions within a protein degradation complex, requiring deletion and single-point recombinant mutants, therefore sparing lengthy preparatory work for the expression and purification of the individual recombinant proteins before having clarified their functional involvement in the degradation complex. PTMe pull-down conditions can also be used for proteomics profiling in the degradation complex discovery phase, but the LC-Ms/Ms approach may not be available to all laboratories. We have successfully used this method towards the characterization of a growth factor–regulated phospho-Degron that we initially identified in an angiogenic/metastatic membrane receptor kinase of the Ephrin family, over-expressed in malignant mesothelioma cells [6]. We have adopted the described protocol in combination with immune-enriched proteomics towards further identifying the protein complex for the degradation of the angiogenic kinase EphB4 [6], whose inhibition by an IGF-II autocrine signal determines its over-expression in a malignant mesothelioma cell line [5].
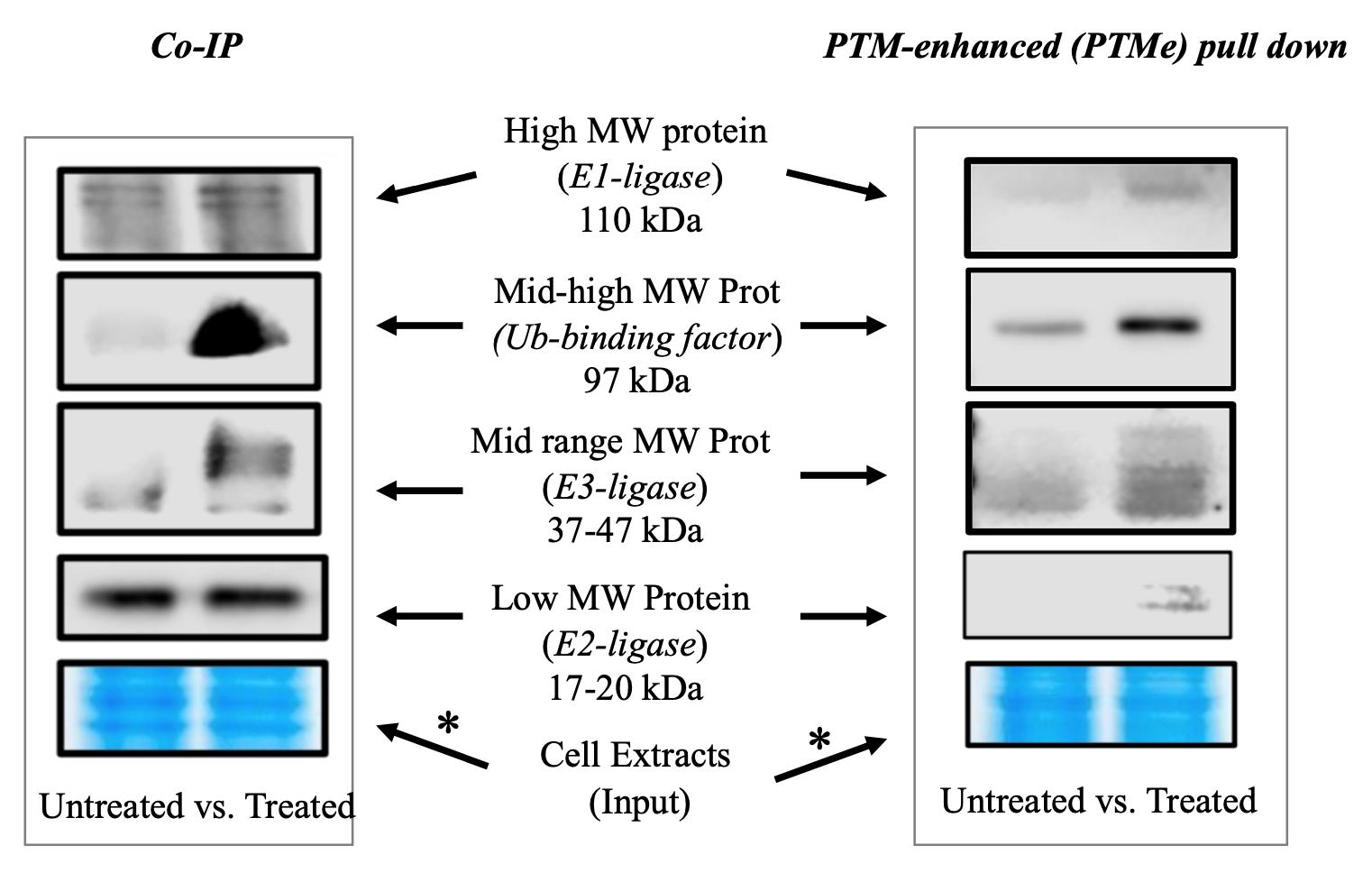
Figure 1. Immuno-blotting detection branch of the post-translational modification–enhanced (PTMe) pull-down method and comparison with traditional co-immune precipitation (co-IP) on the same cellular extract batch from treated vs. untreated cells. An aliquot of the material used in each condition was run on a gel and used for Coomassie stain. The asterisk-marked arrows refer to the complex components for which the method can provide results not observed with traditional co-IP as specified under background. The right-side image is part of the study by Scalia et al. (2023) [6], in which the method was first used. *Please note that two different areas of the same Coomassie gel are shown for comparing the post-normalization protein amount of the same cell extract batches used for both the displayed co-IP and PTMe pull-down experiments.
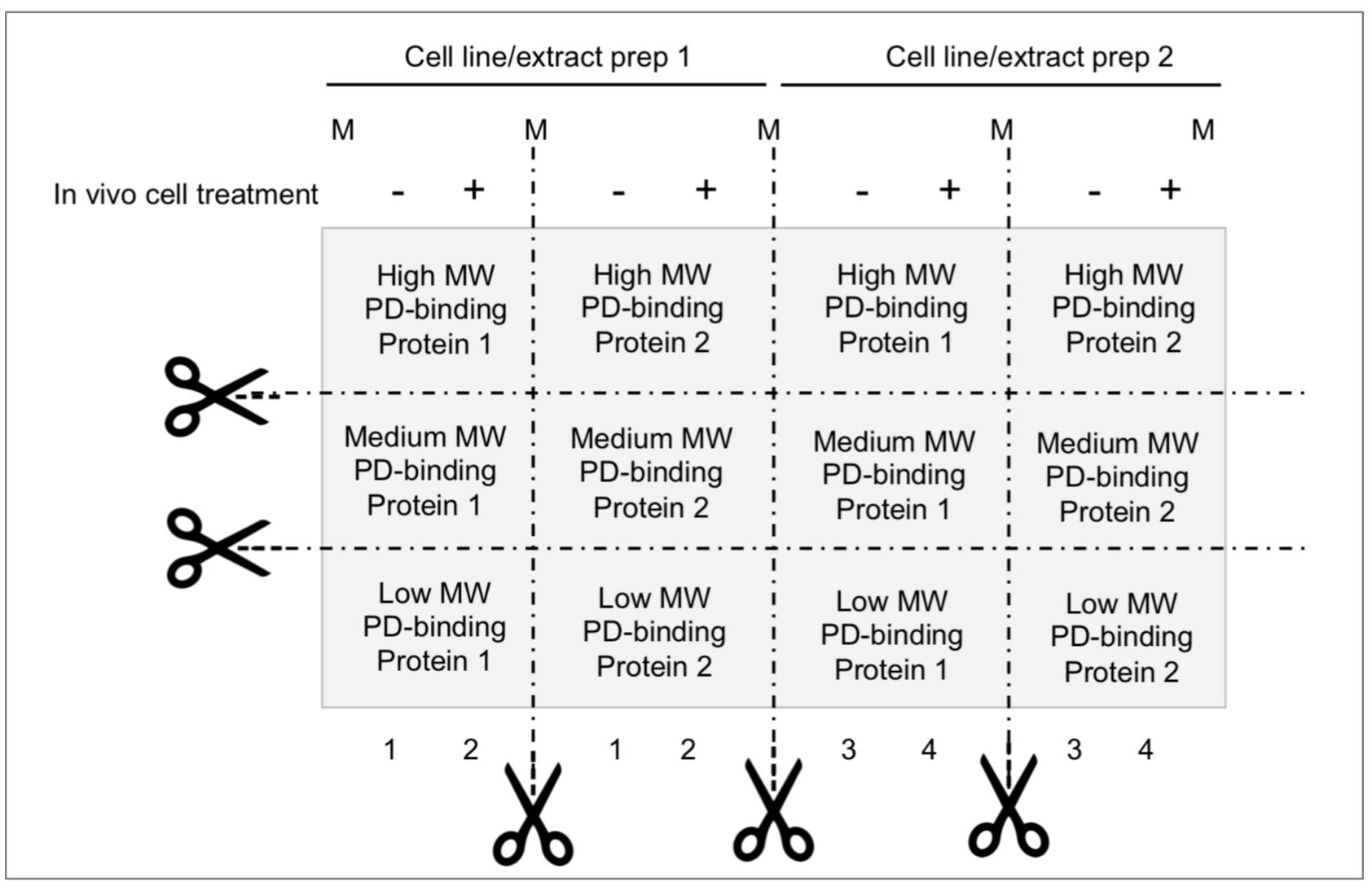
Figure 2. Membrane partition suggested for parallel analysis of ubiquitin (Ub)-E-ligases detection/pDegron recruitment study using post-translational modification–enhanced (PTMe) pull-down assay. Membrane partition is suggested given the relevance of studying same-cell extract batches deriving from individual in vivo treatment conditions and underlying PTMe pulled-down complexes from individual experiment. Proper control suggested between conditions is a Coomassie stain of equal amounts of (protein-normalized) cell extract input resolved by SDS-PAGE.
Materials and reagents
Materials include disposable sterile items like tubing and cell culture supplies obtained from Thermo Inc. (Waltham, MA, USA). Reagents, along with the working concentrations used, have been conveyed in Table 1.
Table 1. Reagents
| Name | Reagent Specs | Working conc. | Provider | Product# |
|---|---|---|---|---|
| Putative PD-synthetic peptide (N/C-biotin tagged) | Assay substrate/bait HPLC-grade | 50 μg/ pull-down reaction (rxn) | New England Peptides (MA) | custom synthesis |
| Ubiquitin, human synthetic | Ub-moiety donor | 1 μg/rxn | Boston Biochemical | U-100H |
| Tris-HCl | pH buffer | 10 mM | Sigma-Aldrich | 77-96-1 |
| PBS | pH buffer | 1× to final rxn vol. | Sigma-Aldrich | D1408 |
| ATP | phosphate moiety donor | 4 mM | Sigma-Aldrich | A1852 |
| DTT | reducing agent | 4 mM | Sigma-Aldrich | D9779 |
| MnCl2 | prot kinase cofactor (Mn++) | 2 mM | Sigma-Aldrich | 1375137 |
| MgCl2 | prot kinase cofactor (Mg++) | 2 mM | Sigma-Aldrich | 1374248 |
| NP40 | Rxn/pull-down facilitator | 0.05% | Sigma-Aldrich | 9036-19-5 |
| D5/W5 inhibitors cocktail | Protease inhibitors | See Recipe 2 | ||
| Kaleidescope prestained protein markers | broad range visible protein markers | 6 µL/lane | Bio-Rad | 1610324 |
| WB-Master Protein Standard | Chemiluminescence Sensitive Markers | 10 µL/lane | Genscript | M00521 |
| SuperSignalTM West Femto Chemiluminescent Substrate | Chemiluminescence Ultrasensitive Reagents system | 0.5 mL/reagent/ Membrane strip | Fisher | 34095 |
| Antigen (/Ab Clone) | Animal source/ clonality | Working concentration | Provider | Clone /product# |
| Ubiquitin (P4D1) | mouse monoclonal | 1:1,000 | Santa Cruz | P4D1/sc8017 |
| Anti-mouse IgG-HRP | chicken monoclonal | 1:2,000 | Santa Cruz | sc2954 |
| Anti-IgGk-LC-HRP | Mouse monoclonal | 1:1,000 | Santa Cruz | sc156102 |
| Streptavidin mag beads | n/a | 2 μL/mL | Genscript | L00936 |
Recipes
The key component of the PTMe pull-down method stands in the homonymous reaction/incubation mixture described below.
PTMe pull-down reaction mixture
PTMe Reaction (rxn) Stock conc. Volume per rxn Final conc. N-Biotin pDegron-peptide coupled w/STP-magnetic beads (see text) n/a 50 μg-STP bound/rxn Tris-HCl, pH 7.5 80 μL 10 mM DTT, 100 mM 8 μL 4 mM ATP, 100 mM 8 μL 4 mM MnCl2,100 mM 4 μL 2 mM MgCl2, 100 mM 4 μL 2 mM Ubiquitin, human 1 µg/µL 1 μL 1 μg Cell extracts (2.5–3.5 µg/µL) Up to 95 μL 0.2–0.3 mg PBS, 0.05% NP40 +D5/W5 protease inhibitors mix
To 200 μL (as needed) 1× Protease and related activities inhibitors stocks
Water soluble inhibitors (W5)
Inhibitor [Stock] µL stock to produce [working]
for 5 mL pull-down buffer[Working] NaVO3 0.2 M 25 µL 1 mM Benzamide 0.25 M 40 µL 2 mM β-glycerophosphate 1 M 50 µL 10 mM NaF 1 M 50 µL 10 mM DTT 1 M 5 µL 1 mM * Sub-total volume (165 µL) - Mol Grade H2O to 10× 483.5 µL n/a PBS, 0.05% NP40 n/a 4,500 µL 1× DMSO soluble inhibitors stock (D5)
Inhibitor [Stock] µL stock to produce [working]
5 mL pull-down buffer
[Working] Pepstatin 10 mg/mL 5 µL 10 µg/mL Leupeptin 10 mg/mL 5 µL 10 µg/mL Aprotonin 10 mg/mL 5 µL 10 µg/mL Okadaic acid 100 µM 5 µL 100 nM PMSF 0.5 M 5 µL 0.5 mM Sub-total volume (25 µL) - DMSO for 10× 487.5 µL n/a PBS, 0.05% NP40 n/a 4,500 µL 1× It is advisable to prepare a larger volume of 10× W5 and D5 stocks, make 0.5 mL aliquots, and save them for single fast thawing (water bath, room temperature). Keep at -20 °C for three months or at -80 °C for six months.
*DTT is added separately to the final buffer containing water-soluble and DMSO-soluble protease inhibitors, to produce a buffer containing 1 mM DTT.
PBST is the antibody probing buffer and is prepared starting by PBS at 1× dilution in sterile deionized H2O with Tween 20 detergent at 0.001% (w/w).
Note: All reagents were purchased from Sigma-Aldrich, St. Louis, MO, USA
Equipment
Cell culture facility (SterilGard III tissue culture hood; CO2 Auto Zero tissue culture incubator, Thermo Scientific, Waltham, MA or comparable models)
4 °C refrigerator or same temperature cold room
-20 °C and -80 °C freezers
Benchtop microcentrifuge supporting 14,000× g spins (e.g., Eppendorf 5418 model or comparable)
Gentle orbital rotation instrument (Boekel Orbital rotator, Feasterville, PA or comparable)
Thermo block with heating capability of 95 °C (Thermo Digital Block Dry Heater, Thermo Scientific, Waltham MA or comparable)
Protein electrophoresis/western blot equipment (NOVEX XCell II and NOVEX TransBlot system, Thermo Scientific, Waltham MA or comparable)
Gel image multi-signal detection system (Odissey XF imaging system, LICOR, Lincoln, NE or comparable). The suggested equipment brands are only indicative and not considered as limiting the outcome of the described protocol
Procedure
文章信息
版权信息
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
如何引用
Scalia, P. and Williams, S. J. (2023). A Post-translational Modification–enhanced Pull-down Method to Study Degron Domains and the Associated Protein Degradation Complexes. Bio-protocol 13(18): e4816. DOI: 10.21769/BioProtoc.4816.
分类
分子生物学 > 蛋白质 > 泛素化
分子生物学 > 蛋白质 > 蛋白质-蛋白质相互作用
分子生物学 > 蛋白质 > 靶向降解
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。