

Protein Structure Predictions, Atomic Model Building, and Validation Using a Cryo-EM Density Map from Hepatitis B Virus Spherical Subviral Particle
利用乙型肝炎病毒球形亚病毒颗粒的低温电子显微镜密度图进行蛋白质结构预测、原子模型构建和验证
(*contributed equally to this work) 发布: 2023年07月20日第13卷第14期 DOI: 10.21769/BioProtoc.4751 浏览次数: 2265
评审: Prashanth N SuravajhalaAnuj KumarHarpreet Singh
参见作者原研究论文
The authors used this protocol in:

Aug 2022
Advertisement








Abstract
Hepatitis B virus (HBV) infection is a global public health concern. During chronic infection, the HBV small-surface antigen is expressed in large excess as non-infectious spherical subviral particles (SVPs), which possess strong immunogenicity. To date, attempts at understanding the structure of HBV spherical SVP have been restricted to 12–30 Å with contradictory conclusions regarding its architecture. We have used cryo-electron microscopy (cryo-EM) and 3D image reconstruction to solve the HBV spherical SVP to 6.3 Å. Here, we present an extended protocol on combining AlphaFold2 prediction with a moderate-resolution cryo-EM density map to build a reliable 3D model. This protocol utilizes multiple software packages that are routinely used in the cryo-EM community. The workflow includes 3D model prediction, model evaluation, rigid-body fitting, flexible fitting, real-space refinement, model validation, and model adjustment. Finally, the described protocol can also be applied to high-resolution cryo-EM datasets (2–4 Å).
Keywords: HBV (HBV)Background
Hepatitis B surface antigen (HBsAg) is both a constituent of the viral envelope and a well-established serological marker for hepatitis B virus (HBV) infection. It exists in three forms (S stands for small, M for middle, and L for large) that share identical C-termini but different N-termini due to in-frame translation from different start codons. Naturally, all three types of HBsAg are incorporated into the envelope of virions in various amounts. Additionally, HBsAg can assemble into non-infectious subviral particles (SVP), either of filamentous or spherical morphology (Heermann et al., 1984; Ganem and Prince, 2004; Gerlich, 2013). The abundance of spherical SVP in hepatitis B carriers is expected to be 1,000–10,000 times higher than that of infectious virions. Despite this, the role of this large surplus of spherical SVP is unknown (Bruns et al., 1998; Chai et al., 2008; Gerlich, 2013; Hu and Liu, 2017). Among these particles, the S-HBsAg is the most abundant protein constituent.
The HBV SVP has many biomedical applications. It is currently used in a licensed vaccine for preventing hepatitis B infection in newborns. When inserted with a foreign antigenic epitope, the chimeric SVP becomes a platform to induce immune response against medically relevant sequences. This method has been used in the development of the malaria vaccine (Guerra Mendoza et al., 2019; Schuerman, 2019; Ho et al., 2020). Furthermore, HBsAg and SVP also possess immunoinhibitory functions (Fang et al., 2015; S. Liu et al., 2015; Tout et al., 2018; Ho et al., 2020; Kim et al., 2020; Megahed et al., 2020). Recently, a new antiviral strategy involving blocking of the assembly and release of SVPs was found to achieve functional control of HBV infection (Mijočević et al., 2019; Vaillant, 2019). Despite having many roles in biomedical applications and hepatitis B infection, there is no detailed structural information on HBsAg or how it assembles into SVP.
To understand the structural relationship between HBsAg and SVP, we purified spherical SVP from patient serum and investigated the structure using cryo-EM. Since the purification steps and cryo-EM structural determination were described elsewhere (H. Liu et al., 2022), this protocol focuses on the procedure for building the atomic model for S-HBsAg by a combination of AlphaFold2 prediction and a 6.3 Å cryo-EM density map of the spherical SVP from HBV genotype E (H. Liu et al., 2022). We began by using AlphaFold2 to predict the 3D models from HBV S-HBsAg sequence. We then evaluated the result of the prediction using a cryo-EM density map and selected the best model for additional analysis. Finally, we combined knowledge from the literature and used flexible fitting to obtain a 3D model for S-HBsAg. This process utilized AlphaFold2, UCSF ChimeraX (Pettersen et al., 2021), ISOLDE (Croll, 2018), PHENIX, MolProbity (Chen et al., 2010), and Coot to generate the Protein Data Bank (PDB) model and to validate the result. This protocol was used to build a 3D model into a moderate-resolution cryo-EM density map (5–7 Å); it can also be applied to experimental data when a high-resolution cryo-EM map (2–4 Å) is available.
Materials and reagents
EMD-26117 (https://www.ebi.ac.uk/emdb/EMD-26117)
HBV S protein sequence (https://www.ncbi.nlm.nih.gov/protein/1035343445)
Equipment
Linux workstation with dedicated GPU or MacBook (with 64 GB RAM).
Software
AlphaFold2 via ColabFold v1.5.2 (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb)
UCSF ChimeraX v1.5 (https://www.cgl.ucsf.edu/chimerax/)
ISOLDE (https://isolde.cimr.cam.ac.uk/)
PHENIX v1.20.1-4487 (https://phenix-online.org/)
MolProbity (http://molprobity.biochem.duke.edu/)
Coot v0.9.8.2 (https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/)
Note: The selection of software for structural modeling is largely a matter of personal preference. ISOLDE, PHENIX, and Coot can all be used for flexible structure modeling. ISOLDE and Coot provide direct visualization during the modeling process, allowing users to make objective decisions by dragging atoms or residues to specific locations. PHENIX, on the other hand, refines structures based on comprehensive checkups. All software can be used individually or in combination for this purpose. However, we have found that using ISOLDE at the beginning provides quick access to make large structural changes, fix Ramachandran and rotamer outliers, and reduce clashes. Additionally, ISOLDE allows users to "pin" part of the protein model at the original location while making new changes to another part of the structure, which is particularly helpful for regions where the density is ambiguous, such as flexible loops. Once an initial model is generated, PHENIX can be used to fine-tune the structure and fix structural outliers, such as residues with poor bond lengths and angles. Finally, we use Coot in a back-and-forth manner with information obtained from MolProbity to refine the final structure until we achieve a satisfactory result (Figure 1).
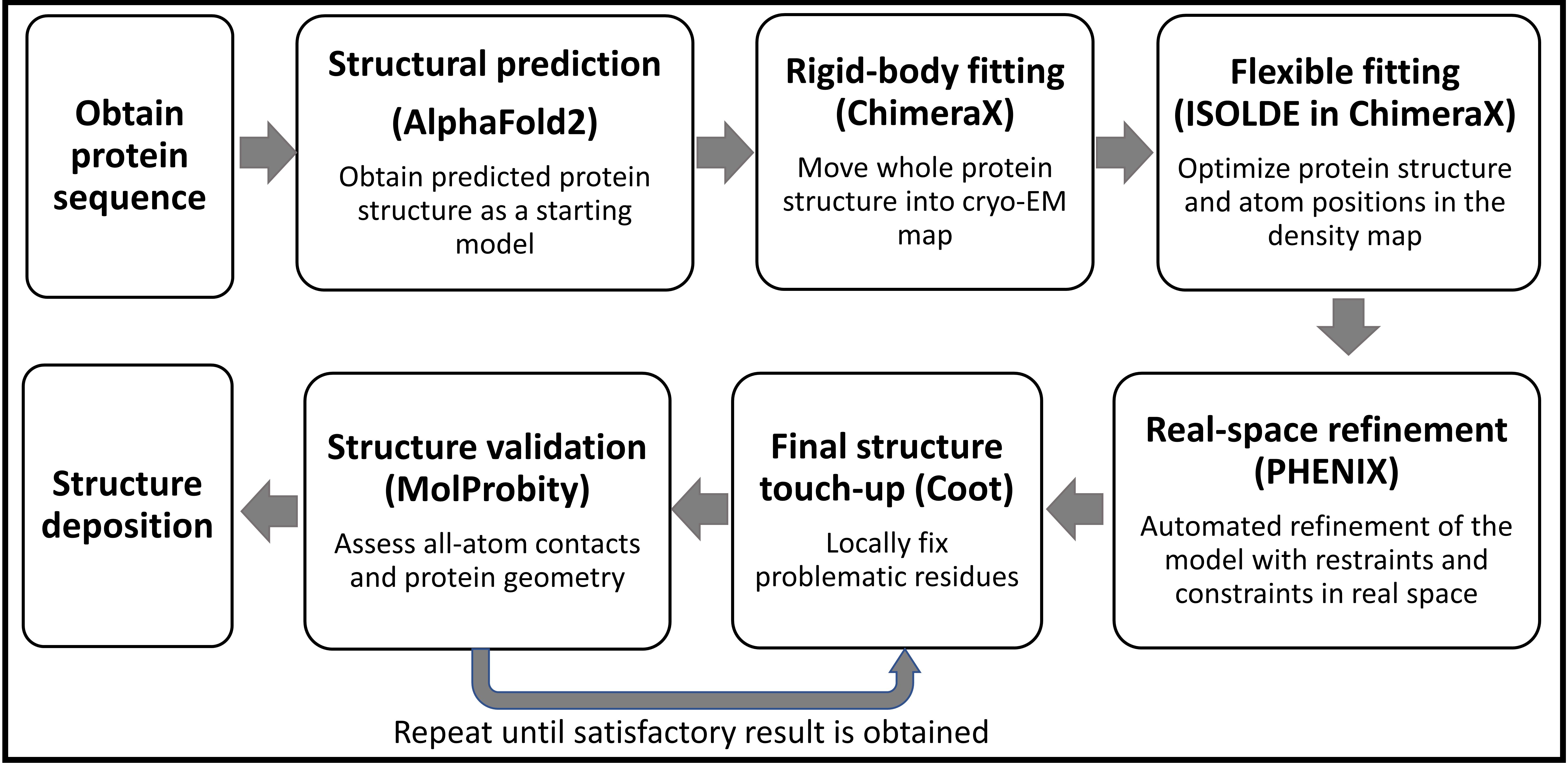
Figure 1. Protocol workflow
Procedure
文章信息
版权信息
© 2023 The Author(s); This is an open access article under the CC BY-NC license (https://creativecommons.org/licenses/by-nc/4.0/).
如何引用
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- DiNunno, N., Bianchini, E. N., Liu, H. and Wang, J. C. (2023). Protein Structure Predictions, Atomic Model Building, and Validation Using a Cryo-EM Density Map from Hepatitis B Virus Spherical Subviral Particle. Bio-protocol 13(14): e4751. DOI: 10.21769/BioProtoc.4751.
- Liu, H., Hong, X., Xi, J., Menne, S., Hu, J. and Wang, J. C. (2022). Cryo-EM structures of human hepatitis B and woodchuck hepatitis virus small spherical subviral particles. Sci Adv 8(31): eabo4184.
分类
微生物学 > 微生物-宿主相互作用 > 病毒
生物物理学 > 电子冷冻断层扫描 > 3D图像重建
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。