

Proteome Integral Solubility Alteration (PISA) Assay in Mammalian Cells for Deep, High-Confidence, and High-Throughput Target Deconvolution
在哺乳动物细胞中进行蛋白质组积分溶解度改变(PISA)测定,用于深度、高置信度和高通量目标解构
发布: 2022年11月20日第12卷第22期 DOI: 10.21769/BioProtoc.4556 浏览次数: 4925
评审: Chiara AmbrogioAnna A. ZorinaAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Feb 2022
Advertisement








Abstract
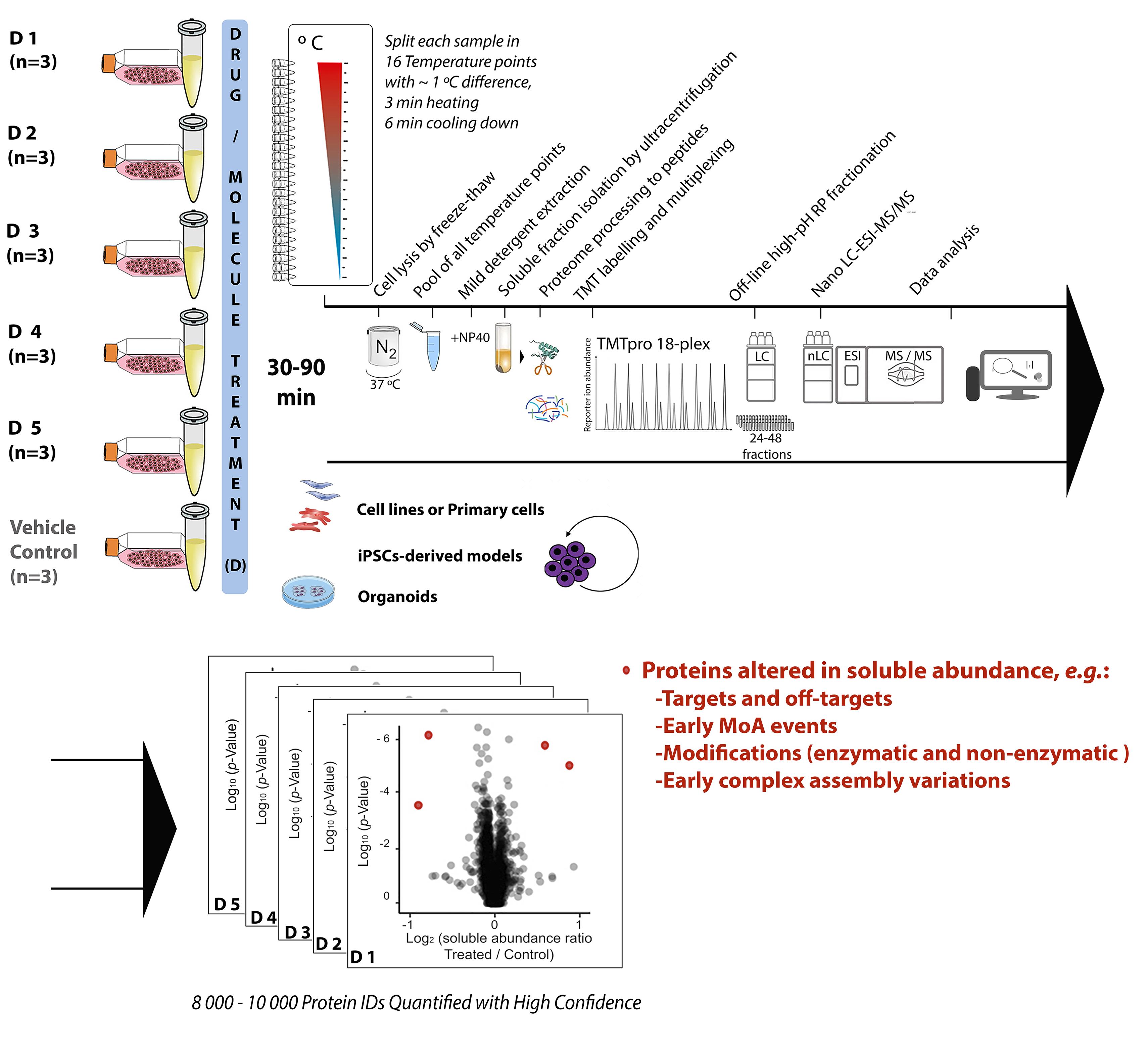
Chemical proteomics focuses on the drug–target–phenotype relationship for target deconvolution and elucidation of the mechanism of action—key and bottleneck in drug development and repurposing. Majorly due to the limits of using chemically modified ligands in affinity-based methods, new, unbiased, proteome-wide, and MS-based chemical proteomics approaches have been developed to perform drug target deconvolution, using full proteome profiling and no chemical modification of the studied ligand. Of note among them, thermal proteome profiling (TPP) aims to identify the target(s) by measuring the difference in melting temperatures between each identified protein in drug-treated versus vehicle-treated samples, with the thermodynamic interpretation of “protein melting” and curve fitting of all quantified proteins, at all temperatures, in each biological replicate. Including TPP, all the other chemical proteomics approaches often fail to provide target deconvolution with sufficient proteome depth, statistical power, throughput, and sustainability, which could hardly fulfill the final purpose of drug development. The proteome integral solubility alteration (PISA) assay provides no thermodynamic interpretation, but a throughput 10–100-fold compared to the other proteomics methods, high sustainability, much lower time of analysis and sample amount requirements, high confidence in results, maximal proteome coverage (~10,000 protein IDs), and up to five drugs / test molecules in one assay, with at least biological triplicates of each treatment. Each drug-treated or vehicle-treated sample is split into many fractions and exposed to a gradient of heat as solubility perturbing agent before being recomposed into one sample; each soluble fraction is isolated, then deep and quantitative proteomics is applied across all samples. The proteins interacting with the tested molecules (targets and off-targets), the activated mechanistic factors, or proteins modified during the treatment show reproducible changes in their soluble amount compared to vehicle-treated controls. As of today, the maximal multiplexing capability is 18 biological samples per PISA assay, which enables statistical robustness and flexible experimental design accommodation for fuller target deconvolution, including integration of orthogonal chemical proteomics methods in one PISA assay. Living cells for studying target engagement in vivo or, alternatively, protein extracts to identify in vitro ligand-interacting proteins can be studied, and the minimal need in sample amount unlocks target deconvolution using primary cells and their derived cultures.
Graphical abstract:
Background
Correct target identification and elucidation of the fine mechanism of action (MoA) of ligands, both covalent and noncovalent, representing research-grade as well as already approved drugs, are both key and major bottlenecks in drug development and drug repurposing (Swinney and Anthony, 2011).
The case of cancer drugs is a clear example of this. It is estimated that 97% of the tested anticancer molecules never make it to the clinic, often because the proposed mechanism of action is incorrect and the observed drug effects are due to off-target toxicity (Lin et al., 2019).
Moreover, the high rate of clinical trial failures clearly indicates the need to seek more precise information on drug targets and the MoA more strictly related to the drug. Therefore, all methodological efforts in this direction are very relevant and worth pursuing. Among the -omics sciences, genomics and transcriptomics have been applied in drug development because of limited operational cost for comparing normal and diseased states, measuring changes in transcription levels, pharmacogenomics, and stratifying patients in clinical trials. However, only mass spectrometry (MS)-based proteomics—and particularly chemical proteomics—can fill in the gap in knowledge of the drug–target–phenotype relationship, by exploring it at a proteome-wide level, with deep proteome coverage, and unbiasedly. Chemical proteomics encompasses different methods, each attacking the problem from a different angle, and each limited by the original intention and experimental design.
The MS-based approaches for the identification of protein interactions using affinity-based purification with chemically engineered ligand probes (Rix and Superti-Furga, 2009) have the strong limitation of the uncertainty in the functional properties of the engineered bait compared to the original ligand. Moreover, even with a well-performing click chemistry used to lock the protein to the bait, the always present promiscuity in binding results in locking background proteins. Additionally, the non-specific noncovalent interactions in the affinity enrichment step lead to an increased background (Wright and Sieber, 2016). The consequent limited specificity of the results called for the development of target deconvolution approaches requiring no chemical modification of the ligand. Therefore, the use of unbiased, proteome-wide, and MS-based proteomics approaches for target deconvolution (Kwon and Karuso, 2018) have recently gained a major place in chemical proteomics.
A promising proteome-wide approach for protein target identification in living cells uses the specificity of protein abundance regulation in ligand-induced late apoptosis (Gaetani and Zubarev, 2019). The concept, originally developed for chemotherapeutics, was also successfully applied to metallodrugs and nanoparticles (Chernobrovkin et al., 2015; Lee et al., 2017; Tarasova et al., 2017), and then extended to a vast library of cancer drugs and multiple cell lines (Saei et al., 2019). This last work produced an online tool assisting in the deconvolution of targets for molecules exhibiting significant cell toxicity at 48h, showing specific regulation of drug targets and mechanistic factors in late apoptosis. However, limitations regarding elucidation of direct primary drug targets and mechanisms direct to the general call for orthogonal methods based on alternative principles.
In thermal proteome profiling (TPP) (Savitski et al., 2014; Franken et al., 2015), drug targets are identified by the increase in protein melting temperatures (Tm) obtained by curve fitting of all temperature points measured in the tandem mass tag (TMT) batch used for each of the drug- or vehicle-treated biological replicates (2 h treatment). Each biological replicate requires a complete TMT batch, and the overall TPP experiment requires as many TMT batches as the biological samples. Therefore, in TPP, scalability and the use of more than two biological replicates per treatment type is complicated by the high sample amounts and costs; straightforward interpretation of the curves obtained within complex intracellular environment as “protein melting” is questionable; data analysis of multibatch TMT and suboptimal curve fitting damage the depth of proteome coverage, with increasing missing values among replicates resulting in false positive and false negative rates (Brenes et al., 2019).
The proteome integral solubility alteration (PISA) assay, commonly named “PISA,” in which this protocol focuses, provides deep and quantitative proteome analysis of the changes in amount of the soluble fraction of each identified protein. Each drug-treated or vehicle-treated sample—thus, each soluble proteome—is first exposed to a gradient of heat as solubility perturbing agent, inducing protein aggregation (Gaetani et al., 2019). Each soluble fraction is isolated, and then deep and quantitative proteomics is applied across all multiplexed biological samples. PISA was proven correct in finding primary and secondary targets in its proof-of-principle work using different drugs with known targets in different cell lines and respective lysates. The method successfully performed target identification and provided elements for MoA elucidation by comparing parallel experiments in living cells and lysates. Indeed, PISA can be applied to living cells for studying target engagement in vivo, or alternatively to protein extracts to identify in vitro ligand-interacting proteins. Further studies applying PISA on an antimicrobial molecule showing anticancer properties were recently published, proving PISA correct also in cases where the target is not known, as it was further validated with follow-up experiments of a different nature (Heppler et al., 2022). Using a statistically relevant number of biological replicates for each tested condition within the same TMT batch, PISA is designed to gain the full robustness and quantification accuracy of TMT multiplexing, and to provide high throughput and high sustainability to target deconvolution, with depth of proteome analysis up to approximately 10,000 proteins in human cells (using current protocols). With up to 18 biological samples per TMT-multiplexed assay, and no thermodynamic interpretation, the PISA assay maximizes throughput, sustainability, proteome coverage, statistical power of analysis (three or more replicates), confidence in results, and several drug molecules analyzed per TMT batch (up to five). The PISA assay conveniently takes advantage of the latest TMTpro technology (Thermo Scientific) (Li et al., 2020b), further extended to 18-plex, allowing for up to nine biological replicates of the ligand compared to vehicle in a single LC–MS/MS experiment. Further developments in TMT technology will immediately increase the PISA throughput. The PISA assay also integrates the advances in deep, quantitative proteomics, including extensive off-line high pH fractionation and nanoscale liquid chromatography (nLC)–MS of all produced fractions.
PISA does away with the sigmoidal solubility-temperature curves and Tm derivation, as each sample is split in equal portions that are exposed to different temperatures before being recomposed into one and multiplexed with the other samples. In PISA, the final amount of each protein would correspond to the integral of a putative solubility-temperature dependence curve, regardless of its shape and function. The information lost on the shape of the solubility-temperature dependence curves turned out to be largely irrelevant for drug target identification. The normalized difference between drug- and vehicle-treated samples is called ΔSm; the ratio (R) of the soluble abundances is also used, and sometimes more advantageous for final data analysis. The number of samples analyzed in PISA by MS is the same regardless of the number of temperature points used, while in TPP, these numbers are in a linear dependence. Therefore, PISA can easily afford a larger number of temperature points, increasing precision of comparative quantification across replicates, which is already increased by removal of the curve fitting. Furthermore, to increase the ΔSm (or R) sensed in PISA by specific targets at a specific temperature range, the last can be narrowed around the region with the highest solubility changes, for most proteins of interest (Li et al., 2020a).
The original PISA assay makes use of the temperature to challenge protein solubility, as temperature is the most optimized and standardized protein aggregation/precipitation agent, easy to control, and removable from samples; however, different PISA assays using organic solvents (Van Vranken et al., 2021) or kosmotropic salts (Beusch et al., 2022) have also been developed. These temperature-free variants of the PISA assay prove its correct design, to robustly interpret and reproducibly measure the proteome amount changes in the soluble fraction as protein solubility rather than protein melting variations.
All in all, PISA increases the throughput by 10–100-fold compared to any other chemical proteomics method, routinely allowing experiments with three or four biological replicates for several compounds simultaneously, and making possible larger studies that would otherwise be prohibitively expensive. Of further relevance, PISA also enables analysis of much reduced sample amounts, and is currently optimized for minimal cell amounts typical for cell culture models of primary cells, iPSC-derived cell cultures, and 3D cell cultures, including organoids.
PISA can also be applied to a range of ligand concentrations, with preferential selection of the target binding at a lower concentration (2D PISA) (Gaetani et al., 2019). In PISA, the reproducible protein amount changes between the soluble fractions of the drug-treated samples and controls can represent the target and off-target interactions, factors involved in early MoA, modified proteins, and early complex assembly variations occurred during the applied drug treatment. The major PISA capabilities of target deconvolution and MoA elucidation reside in the versatility to accommodate within the same TMT multiplex a project design tailored for specific needs, for example, with robust comparisons among various compounds, or incubation at various time points, drug concentrations, or cells. For fuller target deconvolution, PISA can also be integrated in one multiplex with its orthogonal method expression proteomics—altogether named PISA-Express. In PISA-Express, both the proteome regulation as cellular adaptation / response to the drug in 24 h/48 h, together with the solubility shift for 30–90 min incubation in PISA are studied. A relevant feature of PISA-Express is the possibility to analyze solubility alteration at longer times of drug incubation, when cellular protein expression is altered by the drug treatment, and the solubility shifts can be normalized to the total protein abundance variation, due to the integration of PISA and expression proteomics in the same analysis (Sabatier et al., 2021). A third analysis dimension—RedOx proteomics—can also be integrated, and allows the 3D PISA profiling into a single TMT multiplex set, which could be a future industrial standard analysis for drug target identification and MoA elucidation.
Materials and Reagents
Cell culture
Tubes for centrifugation 50 mL and 15 mL (Sarstedt, catalog numbers: 62.559 and 62.554.502)
Sterile 25 cm2 cell culture flask (Sarstedt, catalog number: 83.3910.002)
Cell culture medium [e.g., Dulbecco’s modified Eagle’s medium (DMEM)] (Thermo Fisher Scientific, catalog number: 11685260; original manufacture: Lonza, catalog number: BE12-614F)
Fetal Bovine Serum (FBS, Thermo Fisher Scientific, catalog number: 11560636)
Penicillin-streptomycin solution (Gibco, catalog number: 15140-122)
L-Glutamine (Gibco, catalog number: A2916801)
Ca2+Mg2+-free Dulbecco’s PBS (Cytiva, catalog number: 10462372)
TrypLE express enzyme solution (Thermo Fisher Scientific, catalog number: 12605036)
Dimethyl sulfoxide solution (DMSO, Merck, catalog number: D8418)
PISA treatment
0.2 mL PCR tube strips with caps (Thermo Fisher Scientific, catalog number: AB0452)
Protein low-binding tubes for centrifugation, 1.5 mL and 2 mL (Thermo Fisher Scientific, catalog numbers: 10708704 and 10718894; original manufacture: Eppendorf, catalog number: 0030108116 and 0030108132)
1.5 mL polypropylene tubes for ultracentrifugation (Beckman Coulter, catalog number: 357448)
Ca2+Mg2+-free Dulbecco’s PBS (Cytiva, catalog number: 10462372)
100× protease inhibitor cocktail solution (Thermo Fisher Scientific, catalog number: 78439)
Liquid N2 in a container
Nonidet P-40 (NP40) (Thermo Fisher Scientific, catalog number: 11596671)
Milli-Q water
Cell lysis buffer (see Recipes)
20% NP40 (see Recipes)
Soluble protein fraction processing
Micro-BCA assay kit (Thermo Fisher Scientific, catalog number: 23235)
Dithiothreitol (DTT, Merck, catalog number: D0632)
Iodoacetamide (IAA, Merck, catalog number: I6125)
Acetone (Thermo Fisher Scientific, catalog number: 10417440)
4-(2-hydroxyethyl)-1-piperazinepropanesulfonic acid (EPPS, Merck, catalog number: E9502)
Urea (Merck, catalog number: U5378)
Lyophilized LysC enzyme (FUJIFILM Wako Pure Chemical Corporation, catalog number: 125-05061)
Lyophilized sequencing grade modified trypsin enzyme (Promega, catalog number: V5111)
Trypsin Resuspension Buffer (Promega, catalog number: V5111)
Tandem Mass Tag (TMT) 10-plex, TMTproTM 16-plex, or TMTproTM 18-plex isobaric label reagent set (Thermo Fisher Scientific, catalog numbers: 90110, A44520, or A52045)
Water-free acetonitrile (Thermo Fisher Scientific, catalog number: 10222052)
50% hydroxylamine solution (Thermo Fisher Scientific, catalog number: 90115)
Trifluoroacetic acid (TFA, Merck, catalog number: 302031)
Methanol (Skandinaviska Genetec, catalog number: RH1019/2.5)
Acetonitrile (ACN, Thermo Fisher Scientific, catalog number: A955-212)
Formic acid (FA, Merck, catalog number: 1002641000)
pH indicator paper (VWR, catalog number: 85403.600)
C18 desalting columns (Waters, catalog number: WAT054960)
0.5 M DTT solution (see Recipes)
0.5 M IAA solution (see Recipes)
20 mM EPPS buffer (pH = 8.2) (see Recipes)
20 mM EPPS buffer (pH = 8.2) including 8M urea (see Recipes)
50% ACN solution (see Recipes)
0.1% TFA (v/v) solution (see Recipes)
2% ACN (v/v) solution including 0.1% TFA (see Recipes)
2% ACN (v/v) solution including 0.1% FA (LC-MS buffer A) (see Recipes)
50% ACN (v/v) solution including 0.1% FA (see Recipes)
80% ACN (v/v) solution including 0.1% FA (see Recipes)
Peptide separation, mass spectrometry, and proteomics data analysis
28%–30% NH4OH water solution (Thermo Fisher Scientific, catalog number: 221228-1L-A)
Milli-Q water
Acetonitrile (Thermo Fisher Scientific, catalog number: A955-212)
Reversed-Phase C18 guard column (Waters, catalog number: 186007769)
High pH Reversed-Phase C18 Column (Waters, catalog number: 186003621)
0.1% FA in water (Thermo Fisher Scientific, catalog number: 10188164)
0.1% FA in ACN (Thermo Fisher Scientific, catalog number: 10118464)
Nano trap-column (Thermo Fisher Scientific, catalog number: 164535)
C18 EasySpray nLC peptide separation column (Thermo Fisher Scientific, catalog number: ES803A)
20 mM NH4OH in H2O (high pH Buffer A) (see Recipes)
20 mM NH4OH in ACN (high pH Buffer B) (see Recipes)
98% ACN in H2O including 0.1% FA (LC-MS buffer B) (see Recipes)
Equipment
Cell culture
37 °C 5% CO2 incubator (Thermo Fisher Scientific, Forma Steri-cycle i160)
Laminar flow cabinet (ninoSAFE, class II)
Light microscope (ZEISS, Primo Vert)
Cell counter (Bio-Rad, model: TC10)
Benchtop centrifuge with swinging-bucket rotor for 15 mL tubes (Eppendorf, model: Centrifuge 5804R)
PISA treatment
Thermal cycler (Applied Biosystems, SimpliAmp)
Ultracentrifuge (Beckman Coulter, model: Optima XPN-80)
Fixed-Angle Titanium Rotor for centrifugation (Beckman Coulter, model: Type 45 Ti)
Thermomixer (Eppendorf, model: ThermoMixer C)
Vortex (Scientific Industries, model: Vortex-Genie 2)
Milli-Q water purification system (Millipore, model: IQ 7000)
Soluble protein fraction processing
Absorbance plate reader (BioTek, Epoch)
Benchtop centrifuge for 1.5 mL tubes (Eppendorf, model: Centrifuge 5430R)
Manifold for vacuum extraction for small columns (Waters, Extraction Manifold)
Speed-Vacuum concentrator (Eppendorf, concentrator plus)
Peptide separation, mass spectrometry, and proteomics data analysis
Mass spectrometer equipped with an EASY ElectroSpray source (Thermo Fisher Scientific, Orbitrap Q Exactive HF or higher)
Note: This protocol refers to Orbitrap Q Exactive HF. However, Orbitrap Fusion, Orbitrap Fusion Lumos, Orbitrap Exploris 480, and Orbitrap Eclipse can be also used.
Nanoflow UPLC system with fraction collector and UV detector (Thermo Fisher Scientific, Ultimate 3000)
Capillary HPLC system for peptide high pH C-18 fractionation with fraction collector and UV detector (Thermo Fisher Scientific, Dionex Ultimate 3000)
Software
Absorbance plate reader software (BioTek, Gen5 2.09)
Q Exactive HF-Orbitrap MS 2.9 build 2926 (Thermo Fisher Scientific)
Thermo Scientific SII for Xcalibur (Thermo Fisher Scientific)
MaxQuant software (Max Planck Institute of Biochemistry)
Proteome Discoverer 2.5 (Thermo Fisher Scientific)
Microsoft Office software (Microsoft)
Prism (GraphPad)
Procedure
文章信息
版权信息
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Zhang, X., Lytovchenko, O., Lundström, S. L., Zubarev, R. A. and Gaetani, M. (2022). Proteome Integral Solubility Alteration (PISA) Assay in Mammalian Cells for Deep, High-Confidence, and High-Throughput Target Deconvolution. Bio-protocol 12(22): e4556. DOI: 10.21769/BioProtoc.4556.
- Heppler, L. N., Attarha, S., Persaud, R., Brown, J. I., Wang, P., Petrova, B., Tošić, I., Burton, F. B., Flamand, Y., Walker, S. R., et al. (2022). The antimicrobial drug pyrimethamine inhibits STAT3 transcriptional activity by targeting the enzyme dihydrofolate reductase. J Biol Chem 298(2): 101531.
分类
系统生物学 > 蛋白质组学
分子生物学 > 蛋白质 > 检测
生物化学 > 蛋白质 > 定量
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。