

Whole-genome Methylation Analysis of APOBEC Enzyme-converted DNA (~5 kb) by Nanopore Sequencing
通过纳米孔测序对 APOBEC 酶转化 DNA (~5 kb) 进行全基因组甲基化分析
发布: 2022年03月05日第12卷第5期 DOI: 10.21769/BioProtoc.4345 浏览次数: 7714
评审: Alessandro DidonnaRita Marie Celine MeganckAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Aug 2021
Advertisement










Abstract
In recent years, DNA methylation research has been accelerated by the advent of nanopore sequencers. However, read length has been limited by the constraints of base conversion using the bisulfite method, making analysis of chromatin content difficult. The read length of the previous method combining bisulfite conversion and long-read sequencing was ~1.5 kb, even using targeted PCR. In this study, we have improved read length (~5 kb), by converting unmethylated cytosines to uracils with APOBEC enzymes, to reduce DNA fragmentation. The converted DNA was then sequenced using a PromethION nanopore sequencer. We have also developed a new analysis pipeline that accounts for base conversions, which are not present in conventional nanopore sequencing, as well as errors produced by nanopore sequencing.
Keywords: DNA methylation (DNA甲基化)Background
DNA methylation is an important mechanism for epigenetic regulation of gene expression (Greenberg and Bourc’his, 2019). It has a wide range of effects on genes via several biological processes. DNA methylation is usually detected and analyzed using bisulfite sequencing short reads. However, it is difficult to align these short reads (~150 bp) to some chromosomal regions, such as repetitive sequences and structural variants (Goerner-Potvin and Bourque, 2018). Similarly, short reads also constrain detection of chromosome-specific methylation patterns, such as imprinted regions, in polyploid organisms (Akbari et al., 2021). A comprehensive understanding of epigenetic regulation by DNA methylation will therefore require complementary methods.
The bisulfite method distinguishes between unmethylated and methylated cytosine (C vs. mC), by chemically converting unmethylated C to uracil (U) and to thymine (T), by subsequent amplification (Lister et al., 2009). However, since this reaction is carried out under chemically severe conditions, a large proportion of the DNA in the reaction is fragmented and degraded. The genomic regions that undergo this degradation show biased representation (Olova et al., 2018), further limiting the experimental conclusions this method can provide. The read length of the previous method combining bisulfite conversion and long-read sequencing was only ~1.5 kb, even using targeted PCR (Yan et al., 2015). Recently, enzymatic methyl sequencing (EMseq) was developed as an alternative to the bisulfite method of base conversion (Vaisvila et al., 2021). EMseq involves the oxidation of mC by ten-eleven translocation (TET) enzymes to protect them, followed by base conversion of unmethylated C to U, by APOBEC enzymes. Via the amplification process, U is converted to T, as in the bisulfite method. Because this reaction is performed under milder chemical conditions than with the bisulfite method, longer DNA fragments are obtainable. In fact, a previous study showed that DNA fragments over 5 kb long can be obtained using EMseq and target-specific PCR, and that these fragments can be successfully sequenced in a long-read sequencer (Sun et al., 2021).
Nanopore sequencers read nucleic acid sequences by measuring the change in electric current while the nucleic acids are passing through the nanopore. The maximum read length of nanopore sequencing is over 100 kb (Sakamoto et al., 2020). By recognizing specific electrical patterns for modified bases, base modifications can also be detected (Rand et al., 2017; Simpson et al., 2017). However, while the base-reading accuracy of nanopore sequencers is currently up to 90%, this is not quite high enough to accurately infer methylation patterns (Sakamoto et al., 2020). Furthermore, it requires about 500 ng–1 µg of DNA input, reducing its practical utility for rare samples, such as clinical specimens and biopsies. Although several methods combining base-conversion and long-read sequencing have been developed, thus far all have employed gene-specific amplification (Yang et al., 2015; Liu et al., 2020; Sun et al., 2021). A method for whole-genome methylation analysis by this method, and a bioinformatic pipeline to process the sequence data it generates, have not heretofore been developed.
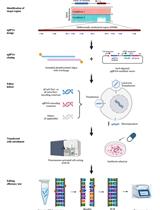
Here, we report a method for whole-genome long-read methylation sequencing, using a relatively small amount of input DNA, for nanopore sequencing of base-converted DNA by APOBEC enzymes (Figure 1) (Sakamoto et al., 2021). Our method, which we designate nanoEM, allows for whole-genome long-read methylation analysis with 10–100 ng of DNA. In addition, we have developed a data analysis pipeline for nanoEM reads by adopting a three-letter alignment approach to long-read alignment. NanoEM is an useful approach for detecting methylation status of structural variants (SVs), repetitive regions, and imprinting regions, which are difficult to analyze using short read sequencing (Sakamoto et al., 2021).

Figure 1. Flow chart of the experimental procedure.
Materials and Reagents
Filter pipette tips 10, 20, 200, and 1,000 µL [e.g., Pipette Tips RT UNV F (RAININ, catalog numbers: 30389172, 30389189, 30389186, and 30389165)]
1.5 mL tubes [e.g., DNA LoBind Tube 1.5 mL (Eppendorf, catalog number: 0030108051)]
PCR tubes [e.g., Temp Assure 0.2 mL PCR 8-Tube Strips, Att. Optical Caps (USA Scientific, catalog number: 1402-4700)]
Mag Attract HMW DNA Kit (QIAGEN, catalog number: 67563)
g-TUBE (Covaris, catalog number: 520079)
Ethanol (e.g., FUJIFILM WAKO Pure Chemical Corporation, catalog number: 057-00456)
Nuclease-free water (Thermo Fisher Scientific, catalog number: AM9930)
Formamide (FUJIFILM Wako Pure Chemical Corporation, catalog number: 064-00423)
NEBNext Enzymatic Methyl-seq kit (New England Biolabs, catalog number: E7120S)
KOD One PCR Master Mix (TOYOBO, catalog number: KMM-101)
Ligation Sequencing kit (Oxford Nanopore Technologies, catalog number: SQK-LSK110)
PromethION flowcell (Oxford Nanopore Technologies, catalog number: FLO-PRO002)
NEBNext Ultra II End Repair/dA-Tailing Module (New England Biolabs, catalog number: E7546)
NEBNext FFPE DNA Repair Mix (New England Biolabs, catalog number: M6630)
NEBNext Quick Ligation Module (New England Biolabs, catalog number: E6056)
Qubit ds DNA HS Assay kit (Thermo Fisher Scientific, catalog number: Q32854)
Agilent DNA 12000 kit (Agilent Technologies, catalog number: 5067-1508)
Agencourt AMPure XP (Beckman Coulter, catalog number: BC-A63880)
DNA Clean & Concentrator-5 (Zymo Research, catalog number: D4013)
ProNex Size-Selective DNA Purification System (Promega, catalog number: NG2001)
TET2 Reaction Buffer with supplement (see Recipes)
70% and 80% (v/v) ethanol (see Recipes)
Wash Buffer of ProNex Size-Selective DNA Purification System (NG2001) (see Recipes)
Equipment
PromethION sequencing device (Oxford Nanopore Technologies, catalog number: PRM48BasicSP)
2100 Bioanalyzer Instrument (Agilent Technologies, catalog number: G2939BA)
Thermal cycler [e.g., T100 thermal cycler (Bio-Rad, catalog number: 1861096)]
Qubit 4 Fluorometer (Thermo Fisher Scientific, catalog number: Q33238)
Vortex Mixer [e.g., Vortex-Genie 2 (Scientific Industries, catalog number: SI-0236)]
High speed centrifuge (e.g., MDX-310 with rack for 2 mL × 24 tubes, TOMY SEIKO)
Tabletop centrifuge for 1.5 and 0.2 mL tubes [e.g., MyFuge mini centrifuge (Benchmark Scientific, catalog number: C1008-B)]
Magnetic stand for 1.5 and 2 mL tubes [e.g., DynaMag-2 (Thermo Fisher Scientific, catalog number: 12321D)]
Magnetic stand for 0.2 mL tubes [e.g., 10× Magnetic Separator (10× Genomics, catalog number: 120250)]
Pipettes for 10, 20, 200, and 1,000 μL tips
Racks for 0.2 mL PCR tubes and 1.5 mL tubes
Software
Python3 (version 3.8.6, https://www.python.org/downloads/)
pysam (version 0.17.0, https://github.com/pysam-developers/pysam)
minimap2 (version 2.22) (Li, 2018)
sambamba (version 0.7.1) (Tarasov et al., 2015)
samtools (version 1.9) (Li et al., 2009)
Integrated Genome Viewer (IGV) (version 2.5.3) (Thorvaldsdóttir et al., 2013)
Procedure
文章信息
版权信息
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Zaha, S., Sakamoto, Y., Nagasawa, S., Sugano, S., Suzuki, A., Suzuki, Y. and Seki, M. (2022). Whole-genome Methylation Analysis of APOBEC Enzyme-converted DNA (~5 kb) by Nanopore Sequencing. Bio-protocol 12(5): e4345. DOI: 10.21769/BioProtoc.4345.
分类
癌症生物学 > 基因组不稳定性及突变
系统生物学 > 表观基因组学 > DNA 甲基化
分子生物学 > DNA > DNA 修饰
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。