

Normalized Ribo-Seq for Quantifying Absolute Global and Specific Changes in Translation
标准化Ribo -Seq 用于量化翻译中的绝对全局和特定变化
(*contributed equally to this work) 发布: 2022年02月20日第12卷第4期 DOI: 10.21769/BioProtoc.4323 浏览次数: 5507
评审: Chiara AmbrogioVaibhav B. ShahAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Mar 2020
Advertisement








Abstract
Ribosome profiling (Ribo-Seq) is a highly sensitive method to quantify ribosome occupancies along individual mRNAs on a genome-wide scale. Hereby, ribosome-protected fragments (= footprints) are generated by nuclease digestion, isolated, and sequenced together with the corresponding randomly fragmented input samples, to determine ribosome densities (RD). For library preparation, equal amounts of total RNA are used. Subsequently, all transcript fragments are subjected to linker ligation, cDNA synthesis, and PCR amplification. Importantly, the number of reads obtained for every transcript in input and footprint samples during sequencing depends on sequencing depth and library size, as well as the relative abundance of the transcript in the sample. However, the information pertaining to the absolute amount of input and footprint sequences is lost during sample preparation, hence ruling out any conclusion whether translation is generally suppressed or activated in one condition over the other. Therefore, the RD fold-changes determined for individual genes do not reflect absolute regulation, but have to be interpreted as relative to bulk mRNA translation. Here, we modified the original ribosome profiling protocol that was first established by Ingolia et al. (2009), by adding small amounts of yeast lysate to the mammalian lysates of interest as a spike-in. This allows us to not only detect changes in the RD of specific transcripts relative to each other, but also to simultaneously measure global differences in RD (normalized ribosome density values) between samples.
Graphic abstract:

Global changes in translation efficiency can be detected with polysome profiling, where the proportion of polysomal ribosomes is interpreted as a proxy for ribosome density (RD) on bulk mRNA. Ribo-Seq measures changes in RD of specific mRNAs relative to bulk mRNA. The addition of a yeast-lysate, as a spike-in for normalization of read counts, allows for an absolute measurement of changes in RD.
Background
Changes in gene expression of protein coding genes are controlled at the level of mRNA transcription and stability, as well as at the level of protein synthesis and degradation (Schwanhausser et al., 2011). While RNA sequencing provides a very sensitive and quantitative means to also detect subtle changes in mRNA transcription and degradation on a global scale, adaptations in nascent and total protein levels, which are commonly measured by mass spectrometric approaches, cannot be quantified as faithfully, due to lower detection and temporal resolution (Dermit et al., 2017). Currently, mostly three different methods are applied to directly measure the rate of newly synthesized proteins by mass spectrometry on a genome-wide scale, including a) pulse-chase stable isotope labeling with amino acids in cell culture (pSILAC) (Schwanhausser et al., 2009), b) SILAC in combination with incorporation of modified amino acids [for example, azidohomoalanine (AHA)] into newly synthesized proteins followed by purification (called BONCAT/QuaNCAT, for quantitative bio-orthogonal non-canonical amino acid tagging) (Dieterich et al., 2007), and c) puromycin incorporation into newly synthesized peptides and purification (PUNCH-P) (Aviner et al., 2014). Alternatively, polysome fractionation in combination with RNA isolation and transcript detection by quantitative real time PCR, microarray analysis, or RNA sequencing has been used to detect changes in the translational status of transcripts. Here, the amount of every transcript per fraction is commonly normalized using equal amounts of spike-in RNA per fraction (Melamed et al., 2009). This approach, however, is either very labor- and/or cost-intensive, or involves the pooling of different polysomal fractions, which obscures especially weak translational changes characterized by small shifts in the polysomal profile. In addition, transcripts with a long coding sequence (CDS) tend to shift less within the polysome profile than those with a short CDS, which distorts the overall analysis. Only with the establishment of ribosome profiling (Ribo-Seq), a highly quantitative and single-codon resolved snap-shot of the current translational status of specific transcripts could be achieved (Ingolia et al., 2009). Unfortunately, with this technique only relative differences in RD of transcripts can be reported, when comparing different samples or conditions. The information regarding absolute changes in the translation rate of a cell is lost. Here, we present a method that we call normalized Ribo-Seq, which allows for the comparison of the relative and absolute occupancies of mRNAs with ribosomes between samples, by normalizing Ribo-seq to a spike-in control. The use of an evolutionarily distant species as spike-in has already been successfully applied to normalize Ribo-Seq datasets in different experimental settings (Cattie et al., 2016; Wang et al., 2018; Haneke et al., 2020; Wang and Gilbert, 2021). For our approach, we add yeast lysate to human lysates as spike-in, after careful quantification by UV absorbance (Figure 1, Note 1). After Ribo-Seq and alignment of sequencing reads to a joint reference of yeast and human transcriptomic sequences, we normalize the number of human ORF-aligned reads to the sum of yeast ORF-aligned reads, to retain information about global changes in RD between conditions.
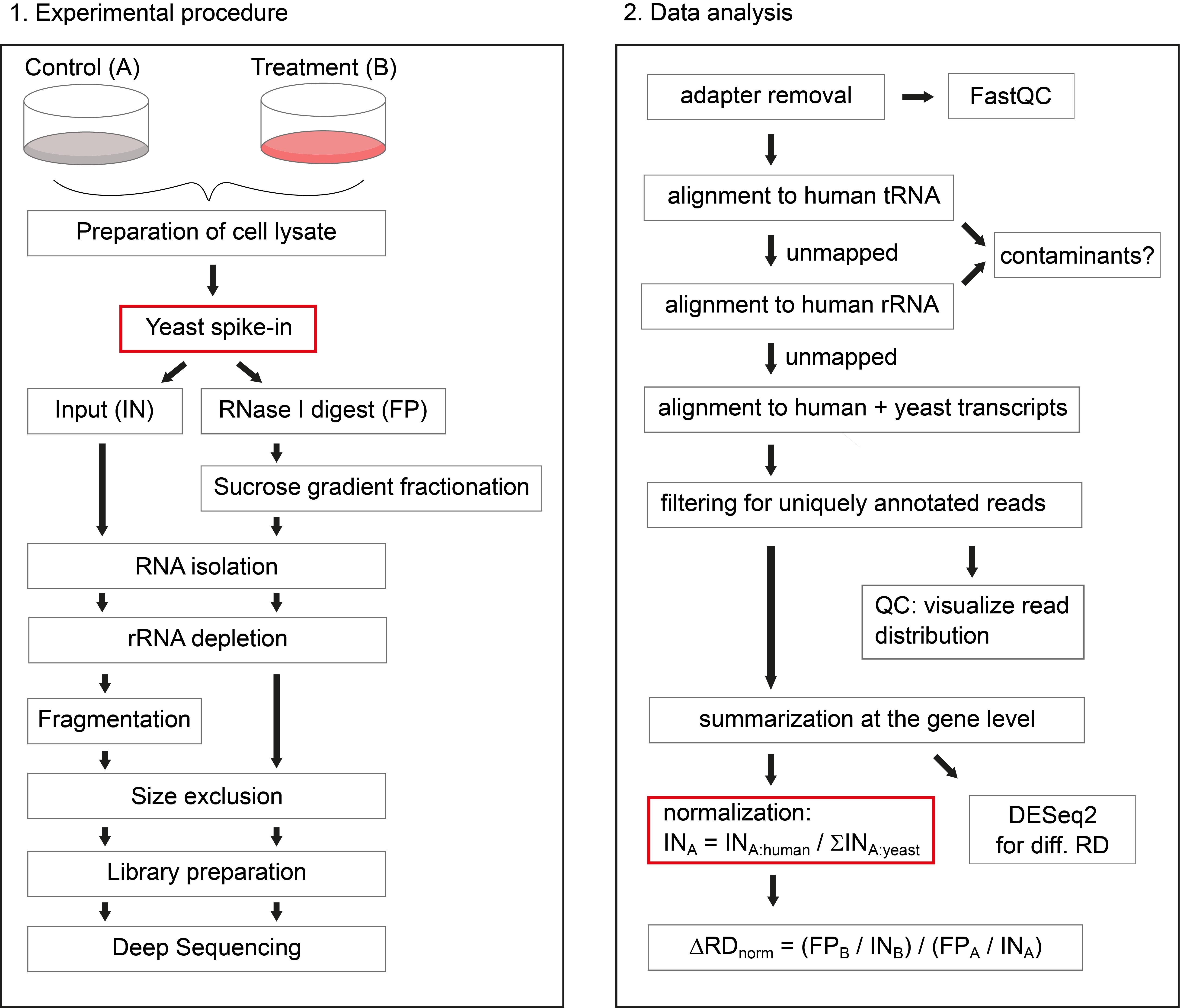
Figure 1. Schematic overview of the protocol. On the left side, the experimental procedure is illustrated, highlighting the addition of yeast lysate as spike-in, after preparation of human cell lysates. On the right side, the data analysis workflow is depicted, highlighting the sample-wise normalization to the sum of yeast ORF reads, to retrieve information about global changes in RD.
Materials and Reagents
Materials
Whole cell Saccharomyces cerevisiae lysate (BY4741 WT strain), flash frozen in liquid nitrogen and stored at -80°C (see Note 2)
10 cm or 15 cm sterile cell culture plates (Sarstedt, catalog numbers: 83.3902 and 83.3903)
Cell lifter (Santa Cruz, catalog number: sc-395252)
Pre-sterilized, RNase-free 1,250 µL, 200 µL, 20 µL, and10 µL micropipette filter tips
Pre-sterilized, RNase-free 0.5 mL, 1.5 mL, and 2 mL microcentrifuge tubes
4 mL open-top thinwall polypropylene tubes 11 × 60 mm (Beckman, catalog number: 328874)
RiboZero Gold Kit for rRNA removal (Illumina, catalog number: MRZG126, see Note 3)
DynaMagTM-2 Magnetic stand (Invitrogen, catalog number: 12321D)
Cutfix stainless scalpel (Braun, catalog number: 5518040)
0.45 µm NanoSep MF tubes (PALL, catalog number: ODM45C34)
21 G syringe needle (Terumo Neolus, catalog number: NN-2150R)
MicroAmp optical 384-well reaction plate (Applied Biosystems, catalog number: 4309849)
MicroAmp optical adhesive film (Applied Biosystems, catalog number: 431971)
Agilent High Sensitivity DNA kit (Agilent, catalog number: 5067-4626)
Agilent small RNA kit (Agilent, catalog number: 5067-1548)
DeNovix dsDNA High Sensitivity kit (Biozym, catalog number: 31DSDNA-HI2)
Qubit microRNA kit (ThermoFisher Scientific, catalog number: Q32880)
NEXTflex small RNA-Seq Kit v3 (Hiss Diagnostics, catalog number: NOVA-5132-06)
NextSeq 500/550 High Output Kit v2.5 (Illumina, catalog number: 20024906)
DMEM (Gibco, catalog number: 41965-039) (in case of RPE1 cells use DMEM/ F12 (Ham’s) medium 1:1 (Gibco, catalog number: 11320-033))
L-glutamine (PAN Biotech, catalog number: P04-80100)
Penicillin/ Streptomycin (PAN Biotech, catalog number: P06-07100)
Fetal bovine serum (FBS, Sigma, catalog number: F7524)
Cycloheximide (CHX, AppliChem, catalog number: A0897)
Nuclease-free water (Ambion, catalog number: AM9937)
Tris
MgCl2
NaCl
KCl
Triton X-100 (Roth, catalog number: 3051.A)
NP-40 (Genaxxon Bioscience, catalog number: M3165)
DTT (AppliChem, catalog number: A2948)
Complete Protease inhibitor, EDTA-free (Roche, catalog number: 11836170001)
Superase Inhibitor (Invitrogen, catalog number: AM2694)
RNase I (Ambion, catalog number: AM2294)
Sucrose
Urea
EDTA
SDS
Phenol:chloroform:isoamylalcohol (PCI, 25:24:1, AppliChem, catalog number: A0944)
Isopropanol
GlycoBlue (15 mg/mL, Ambion, catalog number: AM9515)
Ethanol
NaHCO3
Na2CO3
Glycerol
Bromphenol Blue (AppliChem, catalog number: A2331)
Xylencyanol (AppliChem, catalog number: A4976)
Boric acid
NaOAc
Rotiphorese Gel 30 (37.5:1) polyacrylamide (Roth, catalog number: 3029.1)
Tetramethylethylenediamine (TEMED, AppliChem, catalog number: A1148)
Ammonium peroxodisulfate (APS, AppliChem, catalog number: 131138)
Small RNA ladder (Abnova, catalog number: R0007)
SybrGold (Invitrogen, catalog number: S11494)
RNase OUT (40 U/µL, Invitrogen, catalog number: 10777019)
ATP (Thermo Fisher Scientific, catalog number: R0441)
T4 Polynucleotide kinase (10 U/µL, NEB, catalog number: M0201S) and reaction buffer (10×, NEB, catalog number: B0201S)
SybrGreen Master Mix (Applied Biosystems, catalog number: A25742)
Phosphate buffered saline (PBS) (see Recipes)
Polysome buffer A (2×) (see Recipes)
Polysome buffer B (2×) (see Recipes)
Polysome lysis buffer A (see Recipes)
Polysome lysis buffer B (see Recipes)
Urea buffer (see Recipes)
Random fragmentation buffer (see Recipes)
Stop/Precipitation solution (see Recipes)
Tris-Borate-EDTA (TBE, 10×) (see Recipes)
RNA loading dye (3×) (see Recipes)
25 nt size marker oligo (GCGCGUUACAUCUGGAGUACGAUAC)
35 nt size marker oligo (GGUCCGUCCGGUCUUCUAAUGAACUAGCGAACGAA)
forward primer for test qPCR (GTTCAGAGTTCTACAGTCCGA)
reverse primer for test qPCR (CCTTGGCACCCGAGAATTCCA)
Equipment
Refrigerated tabletop microcentrifuge
End-over-end rotator
Spectrofluorometer (DeNovix, model: DS-11FX+)
Optional: Gradient Master Station (BioComp)
Swinging bucket ultracentrifuge rotor (Hitachi, model: P56ST)
Ultracentrifuge (Hitachi, model: CP80NX)
Continuous UV light monitor and fractionator (Teledyne Isco Foxy Jr)
Fume hood
Thermoblock
Vortex
Thermocycler (Eppendorf Mastercycler)
Mini Protean Tetra Cell polyacrylamide gel box (Bio-Rad) and electrophoresis power supply
Shaker
UV transilluminator or blue light table
Quant Studio Real-Time PCR System (LifeTech, model: 5384)
Bioanalyzer (Agilent, model: 2100 Bioanalyzer)
Sequencer (Illumina, NextSeq500)
Software
We performed all sequencing data analysis steps under Ubuntu 16.04 LTS. The gzip package, perl, and R (with RScript under /usr/bin) must be installed. Depending on the steps that you would like to perform, not all tools might be necessary. For reproducing the analysis exactly as we presented it in our article (Haneke et al., 2020), you can download the directory “tools” from our OSF project page, which contains the versions that we used. For the analysis of new data, it might be preferable to download the newest versions of these tools:
Bowtie (http://bowtie-bio.sourceforge.net/index.shtml) (Langmead et al., 2009)
DESeq2 (http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html) (Love et al., 2014)
FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
fastx (http://hannonlab.cshl.edu/fastx_toolkit/download.html)
PeakTrak v1.10 (Teledyne ISCO, see Note 4)
perl (https://www.perl.org/)
samtools (https://www.htslib.org/)
seqinr (http://seqinr.r-forge.r-project.org/)
sratoolkit (https://github.com/ncbi/sra-tools/wiki/02.-Installing-SRA-Toolkit)
Databases
The code shown in this protocol, our in-house developed scripts, and further required files are available from our OSF project page. Other versions of tRNA, rRNA, transcriptomic, and genomic sequences can be downloaded from the Table Browser of the UCSC Genome Browser. Unprocessed read sequences of our own study are available from GEO via SRA (see Data Analysis, D):
UCSC Genome Browser (https://genome.ucsc.edu/)
OSF (https://osf.io/wr23s/?view_only=1f6fafb4f1d144f8a6093730ebe50f2a)
GEO (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128538)
Procedure
文章信息
版权信息
© 2022 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Hoerth, K., Reitter, S. and Schott, J. (2022). Normalized Ribo-Seq for Quantifying Absolute Global and Specific Changes in Translation. Bio-protocol 12(4): e4323. DOI: 10.21769/BioProtoc.4323.
- Haneke, K., Schott, J., Lindner, D., Hollensen, A. K., Damgaard, C. K., Mongis, C., Knop, M., Palm, W., Ruggieri, A. and Stoecklin, G. (2020). CDK1 couples proliferation with protein synthesis. J Cell Biol 219(3): e201906147.
分类
分子生物学 > RNA > RNA-蛋白质相互作用
生物化学 > RNA > mRNA翻译
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。