

Protocol for RNA-seq Expression Analysis in Yeast
酵母RNA-seq表达分析方法
发布: 2021年09月20日第11卷第18期 DOI: 10.21769/BioProtoc.4161 浏览次数: 6154
评审: Shyam SolankiJihyun KimAnonymous reviewer(s)

参见作者原研究论文
The authors used this protocol in:

Dec 2020
Advertisement









Abstract
Genome-wide sequencing of RNA (RNA-seq) has become an inexpensive tool to gain key insights into cellular and disease mechanisms. Sample preparation and sequencing are streamlined and allow the acquisition of hundreds of gene expression profiles in a few days; however, in particular, data processing, curation, and analysis involve numerous steps that can be overwhelming to non-experts. Here, the sample preparation, sequencing, and data processing workflow for RNA-seq expression analysis in yeast is described. While this protocol covers only a small portion of the RNA-seq landscape, the principal workflow common to such experiments is described, allowing the reader to adapt the protocol where necessary.
Graphic abstract:
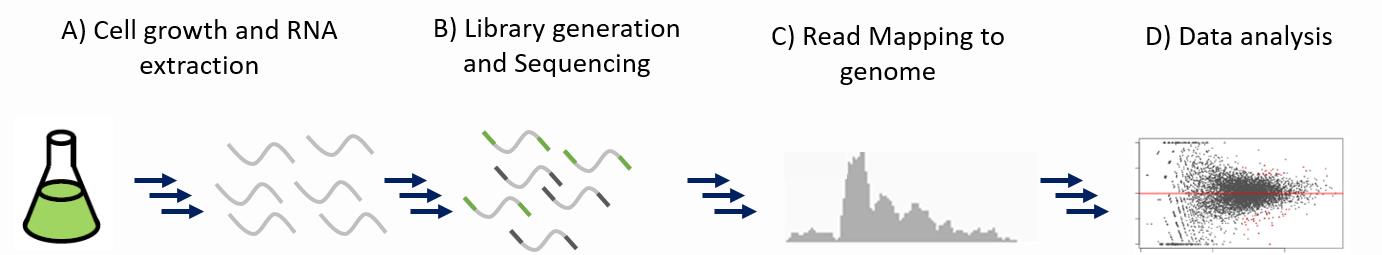
Basic workflow of RNA-seq expression analysis.
Background
Sequencing of RNA (RNA-seq) has – with the emergence of next-generation sequencing – become a powerful tool to measure the presence and quantity of RNA in a given cell population or even within a single cell. Since its initial uses (Bainbridge et al., 2006; Cheung et al., 2006; Emrich et al., 2007; Weber et al., 2007; Nagalakshmi et al., 2008), RNA-seq has seen a large variety of applications: from gene expression analysis by quantitating the relative amounts of RNA sequence reads to the discovery of novel transcripts or splice variants, ribosome profiling, or the detection of single nucleotide polymorphisms. Even though a manifold of specific RNA-seq uses exists, the basic workflow remains the same: RNA molecules are extracted, amplified, sequenced, aligned to the genome of the host organism, and, subsequently, the data is analyzed. Sample requirements are relatively low; typically, 1 μg down to 10 ng input RNA is sufficient for downstream amplification and library generation. For single-cell RNA-seq, as little as 10 pg is required since the low amount of input material is amplified prior to library generation (Haque et al., 2017). RNA-seq can be applied to any population of extracted RNA, independent of the source organism. Due to the wide applicability of RNA-seq, sample preparation kits that vary in complexity are commercially available (from RNA extraction to whole RNA-seq library generation, including the computational analysis).
An integral part of RNA-seq is sequencing of the extracted RNA population. Most commonly, sequencing is performed by the detection of fluorescently labeled nucleic acids bound to the surface of flowcells, e.g., using platforms such as Illumina and PAC Biosystem. To this end, the RNA fragments are converted into a cDNA library and amplified, and flowcell adapters are introduced. During each sequencing cycle, DNA polymerases attach fluorescently labeled nucleotides to the flowcell-bound library molecules, which are then detected by the sequencer, typically generating read lengths of 150–300 bp to several Kbp (for Illumina and PAC Biosystem, respectively). More recently emerging is sequencing by passage of nucleic acids through protein nanopores embedded in membranes (e.g., by Nanoporetech) (Logsdon et al., 2020), allowing for the sequencing of much longer fragments (up to Mbp). At the time of writing, the most commonly available sequencers (e.g., Illumina or PAC Biosystem) cost around $100 k for the instrument alone, whereas table-top sequencers using the nanopore technology are considerably cheaper (~$10 k), promising wider applicability in the near future. Due to the considerable cost of the most commonly available sequencing systems, resources are often shared among labs or institutes and managed by trained professionals that ensure the acquisition and integrity of high-quality sequencing data.
While it extends beyond the scope of this manuscript to describe all the applications of RNA-seq, this protocol aims to provide a workflow for RNA-seq expression analysis that can be used as a reference backbone, which the reader can adapt to their specific needs (e.g., RNA extraction from a different source or the addition of splice-aware alignment steps for genomes of higher eukaryotes). RNA-seq expression analysis is a powerful and commonly used tool to identify genes that are up- or downregulated in a stressed sample (e.g., in the presence of genomic mutations, UV light, drugs, chemical or nutrient stress) as compared with a relaxed sample (e.g., wild-type cell population). A gene is “upregulated” or “downregulated,” respectively, when more or less of its RNA is measured (i.e., expressed in the cell) under the stressed conditions as compared with the wild type.
Here, the workflow for RNA-seq expression analysis in S. cerevisiae is described, from cell growth to RNA extraction, library generation, data processing, and analysis. This protocol focuses on using commonly available lab resources wherever possible and utilizes open source and free-of-cost software packages provided by the bioinformatics community. This workflow has proven to be robust and useful for the analysis of gene expression profiles in libraries of histone point mutants in yeast (Braberg et al., 2020).
Materials and Reagents
1.5 ml Eppendorf tubes (e.g., Eppendorf, catalog number: 0030120086)
Petri dishes, plastic, 10-cm diameter (e.g., Falcon, catalog number: 353003)
Sterile pipette tips
Toothpicks (autoclaved)
Dry ice
Agar (e.g., Becton Dickinson, catalog number: 214030)
Bacto Peptone (e.g., Becton Dickinson, catalog number: 211677)
CHCl3 (Acid phenol, e.g., ThermoFisher, catalog number: AM9720)
DEPC-ddH2O (Diethyl pyrocarbonate-treated water, e.g., Invitrogen, catalog number: 750024)
EDTA (Ethylenediaminetetraacetic acid disodium salt dihydrate, e.g., Sigma-Aldrich, catalog number: E6635)
EtOH (Ethanol, e.g., Sigma-Aldrich, catalog number: 459836)
Formamide (e.g., Sigma-Aldrich, catalog number: 11814320001)
Glucose (e.g., Molekula, catalog number: 13002238)
HCl (Hydrochloric acid, e.g., Sigma-Aldrich, catalog number: 320331)
NaAc (Anhydrous sodium acetate, e.g., Sigma-Aldrich, catalog number: S2889)
NaOH (Sodium hydroxide pellets, e.g., Sigma-Aldrich, catalog number: 1064980500)
SDS (Dodecyl sulfate sodium salt, e.g., Merck, catalog number: 13760)
Tris (2-Amino-2-(hydroxymethyl)-1,3-propanediol, e.g., Sigma-Aldrich, catalog number: T1503)
Yeast extract (e.g., Serva, catalog number: 24540)
Yeast Extract Peptone Dextrose (YEPD) media (see Recipes)
1 M Tris-HCl solution, pH 7.5 (see Recipes)
0.5 M EDTA solution, pH 8.0 (see Recipes)
20% SDS solution (see Recipes)
Tris-EDTA-SDS (TES) solution (see Recipes)
3 M NaAc solution, pH 5.2 (see Recipes)
Equipment
Autoclave
Centrifuge and table-top centrifuge
Vortex
Flasks, autoclavable
Incubator
pH meter
Pipettes (1-ml, 200-μl, 20-μl, 2-μl)
Stir bar and stir plate, magnetic
Thermocycler
Software
bbmap (BBMap – Bushnell, B.; Version 38.90; sourceforge.net/projects/bbmap)
Biocmanager (https://cran.r-project.org/web/packages/BiocManager/vignettes/BiocManager.html; Version 3.12; https://bioconductor.org/install/)
bioconda (Grüning et al., 2018; http://bioconda.github.io/user/install.html)
bowtie2 (Langmead and Salzberg, 2012; Version 2.4.2; http://bowtie-bio.sourceforge.net/bowtie2/index.shtml)
bwa (Burrows-Wheeler Aligner, Li and Durbin, 2009; Version 0.7.17; http://bio-bwa.sourceforge.net/)
DESeq2 (Love et al., 2014; Version 1.30.1, http://bioconductor.org/packages/release/bioc/html/DESeq2.html)
fastqc (Andrews, 2010; FastQC: a quality control tool for high throughput sequence data; Version 0.11.9; https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
htseq-count (Anders et al., 2014; Version 0.11.1; https://htseq.readthedocs.io/en/release_0.11.1/install.html)
Integrated Genomics Viewer (Robinson et al., 2011; Version 2.9.2; https://software.broadinstitute.org/software/igv/download)
R (R Core Team, 2017; Version 4.0.4; https://cran.r-project.org/bin/windows/base)
samtools (Li et al., 2009; Version 1.11; http://www.htslib.org/download/)
tophat (Langmead et al., 2009; Version 2.1.1; https://ccb.jhu.edu/software/tophat/index.shtml)
trimmomatic (Bolger et al., 2014; Version 0.40; http://www.usadellab.org/cms/?page=trimmomatic)
Procedure
文章信息
版权信息
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Bohn, S. (2021). Protocol for RNA-seq Expression Analysis in Yeast. Bio-protocol 11(18): e4161. DOI: 10.21769/BioProtoc.4161.
- Braberg, H., Echeverria, I., Bohn, S., Cimermancic, P., Shiver, A., Alexander, R., Xu, J., Shales, M., Dronamraju, R., Jiang, S., Dwivedi, G., Bogdanoff, D., Chaung, K. K., Huttenhain, R., Wang, S., Mavor, D., Pellarin, R., Schneidman, D., Bader, J. S., Fraser, J. S., Morris, J., Haber, J. E., Strahl, B. D., Gross, C. A., Dai, J., Boeke, J. D., Sali, A. and Krogan, N. J. (2020). Genetic interaction mapping informs integrative structure determination of protein complexes. Science 370(6522).
分类
系统生物学 > 基因组学 > 测序
微生物学 > 微生物生物化学 > RNA
分子生物学 > RNA > RNA 检测
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。