

Preparation of Drosophila Larval Blood Cells for Single-cell RNA Sequencing
果蝇幼虫血细胞单细胞RNA测序的制备

发布: 2021年08月20日第11卷第16期 DOI: 10.21769/BioProtoc.4127 浏览次数: 3779
评审: Gal HaimovichHongjie Li

参见作者原研究论文
The authors used this protocol in:

May 2020
Advertisement








Abstract
Recent advances in single-cell RNA-sequencing (scRNA-seq) technologies provide unprecedented opportunities to identify new cell types and characterize cell states. One of the most important requirements for performing scRNA-seq is to obtain high-quality single cells in suspension. Recently, we used this approach to characterize Drosophila blood cells (hemocytes). Here, we provide a detailed protocol for obtaining single hemocytes in suspension, which can be used for microfluidics-based scRNA-seq platforms. This protocol involves the simple bleeding of third instar larvae and the subsequent purification of the hemolymph using either Optiprep-based gradient centrifugation or traditional centrifugation methods to obtain single hemocytes of high quality for scRNA-seq. Importantly, this method for single-hemocyte preparation is straightforward and reproducible, with negligible issues associated with cell viability as the entire procedure involves no enzymatic dissociation.
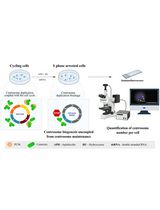
Graphic abstract:
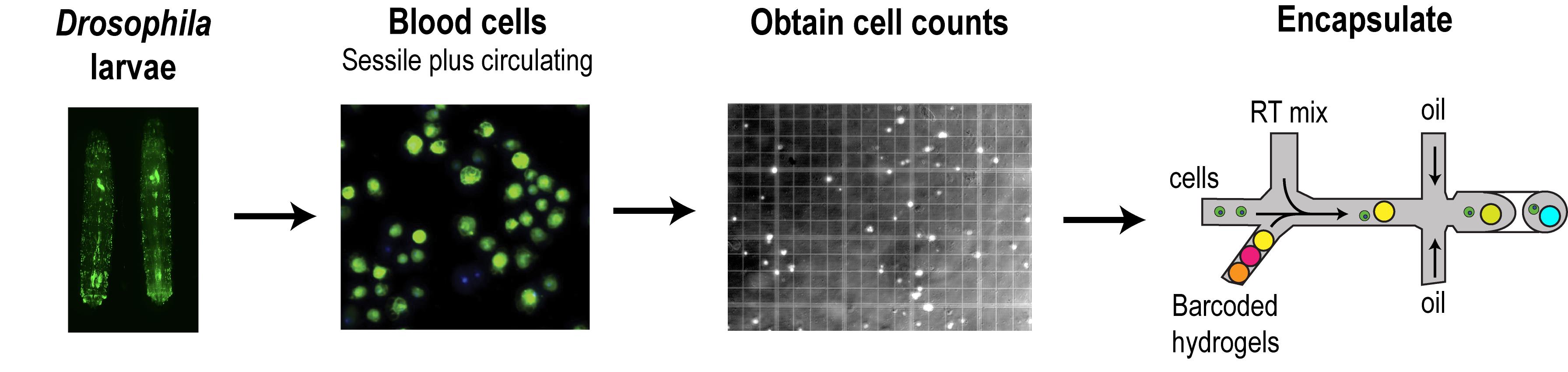
Workflow for the preparation of Drosophila larval blood cells in suspension. Hemocytes (blood cells) of the sessile and circulatory compartments of larvae are derived by simple bleeding and purification using gradient centrifugation. Blood cells are counted and subsequently encapsulated by microfluidics-based scRNA-seq platforms. Blood cells represented in the schematic are derived from third instar larvae of the genotype Hemolectin-GAL4.Delta, UAS-2xEGFP (BDSC stock #30140).
Background
Drosophila myeloid-like blood cells, also called hemocytes, are composed of three major types: plasmatocytes, crystal cells, and lamellocytes, which are analogous to macrophages, platelets, and giant cells, respectively, in higher vertebrates (Banerjee et al., 2019). These hemocytes are highly dynamic in nature and respond to various inflammatory conditions, thus making Drosophila a simple and ideal genetic model to study hematopoiesis, infections, wounding, and other blood disorders (Vlisidou and Wood, 2015; Banerjee et al., 2019). Hemocytes are mainly derived from embryonic and larval hematopoiesis, and are primarily located as sub-epidermal ‘hubs’ (otherwise known as sessile/resident hemocyte populations), with a fraction of hemocytes circulating in the larval hemolymph (Markus et al., 2009; Gold and Bruckner, 2015). The sessile hemocytes can be mechanically dislodged and forced into circulation for a brief period by applying a force to the larvae, such as by vortexing (Petraki et al., 2015). This “hemocyte disturbance assay” allows for the retrieval of blood cells in higher numbers, which typically cannot be obtained by larval bleeding without prior perturbation, and thus is useful for experimental analyses requiring significant cell material. In cases where circulating and sessile hemocyte populations need to be assayed separately, a “hemocyte bleed/scrape assay” could be employed, in which circulating cells can be bled and obtained prior to obtaining sessile populations by the ‘scraping’ method. For a detailed experimental approach to the larval bleeding method in general, we highly recommend the visual protocol by Brückner and colleagues (Petraki et al., 2015). We have adapted the “hemocyte disturbance assay” as one of the critical steps in our approach (Tattikota et al., 2020) to isolate and prepare circulating hemocytes together with sessile hemocytes for their subsequent use in microfluidics-based scRNA-seq platforms, including inDrops (Klein et al., 2015; Zilionis et al., 2017), Drop-seq (Macosko et al., 2015), and 10X Genomics (Zheng et al., 2017). By combining mechanical disruption, simple bleeding, and gradient or traditional centrifugation, we obtain single hemocytes in suspension with a higher yield of cells without compromising cell quality. Since this protocol does not involve enzymatic dissociation, isolated hemocytes are of high quality with regards to viability and are therefore advantageous for downstream scRNA-seq methodologies.
Materials and Reagents
Ice bucket with ice
Distilled water
Small paintbrush, round, size 2 (Princeton) or a spatula to collect larvae
PYREX 9-cavity Spot Plates (Fisher Scientific, catalog number: 13-748B)
Pipettes and tips (P200, P1000)
Glass beads, unwashed, 425-600 μm (Sigma, catalog number: G9268-100G), store at RT
2-ml Safe-Lock Eppendorf tubes (VWR Scientific, Eppendorf, catalog number: 20901-540)
100-μm cell strainer (VWR Scientific, catalog number: 21008-950)
Eppendorf LoBind tubes (Fisher Scientific, Eppendorf, catalog number: 13-698-794)
Eppendorf microcentrifuge tubes (VWR, catalog number: 62111-752)
0.22-μm 50-ml tube top filter (VWR Scientific, catalog number: 28680-120)
Petri dishes, 60 mm (VWR Scientific, catalog number: 25384-060)
15-ml Falcon tubes (Corning, catalog number: 430052)
Trypan Blue solution (Sigma, catalog number: T8154), store at RT
Neubauer counting chamber (hemacytometer) (VWR Scientific, catalog number: 15170-172)
OptiPrep (Axis-Shield, catalog number: AXS-1114542), store in a dark place at RT
1× phosphate-buffered saline (ThermoFisher, catalog number: 10010049), store at RT
Schneider’s insect media (Sigma-Aldrich, catalog number: S9895), store at 4°C
Glycerol (Sigma-Aldrich, catalog number: G7757), store at RT
Bovine serum albumin (BSA) (Sigma, catalog number: A3912), store at 4°C
Optiprep gradient solution (see Recipes)
0.5% BSA in 1× PBS (see Recipes)
20% glycerol solution (see Recipes)
Equipment
#5 tweezers (two pairs) (World Precision Instruments, Dumont, catalog number: 501985)
Cold centrifuge with swinging bucket rotor set to 4°C or a cold benchtop centrifuge
Stereomicroscope (for dissecting larvae)
Light microscope (for manual counting of cells)
(Optional) Countess 3 automated cell counter (Thermo Fisher Scientific)
Procedure
Timeline
Expect the following time in minutes [min] before you reach the cell encapsulation step.
Larvae collection and clean-up: ~30 min
Transferring to Eppendorf tubes and vortexing: ~3 min
Dissection/bleeding: ~15 min
Optiprep spin including “no brake”: ~30 min
Cell recovery and counting: ~10-15 min
The total time is ~90 min-2 h for two samples with 100 larvae per genotype or condition.
Prepare Optiprep solution
To achieve a concentration of 1.09 g/ml, simply mix Optiprep and PBS at a ratio of 0.27 (Optiprep): 0.73 (PBS). That is, for 2 ml, mix 540 μl optiprep with 1460 μl PBS. Mix thoroughly in a 15-ml Falcon tube and place on ice.
Stepwise protocol to obtain high-quality hemocytes from third instar larvae
Collect ~100-200 precisely timed (96 h or 120 h after egg laying) larvae for each condition in PBS. Larvae can easily be retrieved from fly vials using 20% glycerol solution. Add generous volumes of 20% glycerol to the fly vials containing the larvae in food; mix the food with a spatula by stirring. Allow the larvae to float and retrieve them using a paintbrush or small spatula (Figure 1A).
Wash the larvae briefly using ~300 μl water or PBS in a 9-well chambered glass plate, and using a paintbrush, load all of them into 2-ml Eppendorf tubes containing ~500 μl glass beads and ~200 μl PBS or water (Figure 1B).
Vortex the tubes containing the larvae and glass beads for 1.5-2 min at high speed.
Decant the larvae and beads (you may have to use additional water to force the larvae and beads from the tubes) into a small Petri dish (Figure 1C). Collect the larvae, and using a paintbrush, transfer to a 9-well chambered glass plate containing 200 μl PBS or Schneider’s media in one of the wells (Figure 1D). The chambered glass plate can be placed on ice.
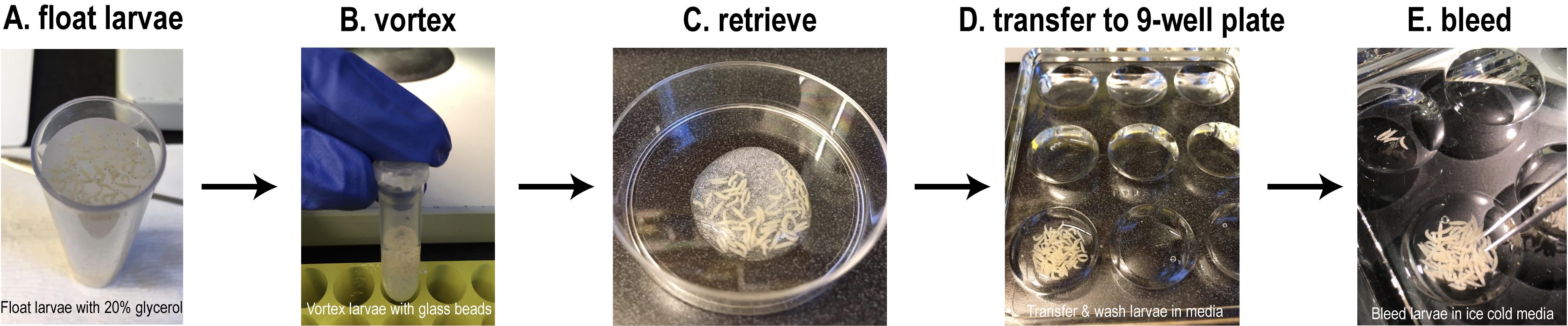
Figure 1. Experimental setup for bleeding Drosophila third instar larvae. A. Retrieve the larvae from fly food using 20% glycerol. B. Wash larvae briefly, transfer to an Eppendorf tube containing glass beads, and vortex. C. Decant the vortexed larvae into a Petri dish. D. Transfer the larvae to a 9-well chambered glass plate containing PBS or Schneider’s media for a brief wash on ice. E. Transfer the washed larvae to another well containing ice-cold Schneider’s media or PBS containing 0.5% BSA and bleed the larvae using #5 tweezers under a stereomicroscope. See Figure 2 for more details.Next, transfer the larvae into another well containing 200 μl ‘ice cold’ Schneider's media or PBS containing 0.5% BSA (Figure 1E). Move the plate under a stereomicroscope with a light source to visualize the larvae.
While observing the larvae under the stereomicroscope, hold the posterior end of one larva with one pair of tweezers and use the other pair to gently nick open the posterior end and continue tearing toward the anterior end (Figure 2). Repeat the procedure for all larvae. Allow the larvae to bleed for at least one minute.

Figure 2. Schematic for bleeding Drosophila third instar larvae. Tweezer #1 is used to hold the posterior end of each larva, while Tweezer #2 is used to tear the larvae in the anterior direction. The bleeding releases the hemolymph containing the hemocytes into the well containing PBS/Schneider’s media.Gently remove the PBS/BSA or Schneider’s media containing the blood cells using a P200 pipette. Filter the cell suspension directly into a clean Eppendorf LoBind microcentrifuge tube on ice using a 100-μm cell strainer to remove any chunks or debris.
Next, gently overlay 200 μl filtered cell suspension onto 2 ml 1.09 g/ml Optiprep gradient previously prepared in a 15-ml Falcon tube on ice.
Proceed to the next condition of larvae (if applicable), obtain the cell suspension (Steps B1-B8), and overlay onto another Optiprep gradient in the second 15-ml Falcon tube on ice.
Note: Ideally, the two conditions or genotypes of larvae are vortexed simultaneously and bled one after the other into separate wells of the glass plate; the cell suspensions are then loaded onto two different Optiprep gradient tubes, which may save time.
Spin the Falcon tubes with cells over Optiprep at 1,000 × g in a swinging bucket rotor set to 4°C for 20-30 min with the ‘no brake’ setting.
Carefully collect the cells from the gradient. First, collect the top 100 μl that you loaded and transfer to a clean Eppendorf LoBind tube. It is recommended to collect an additional 100 μl from the top and transfer to another clean Eppendorf LoBind tube. Count the cells from both tubes to see which contains more cells. Sometimes, the cells accumulate in the second 100 μl region of the gradient.
Count the cells using a Neubauer counting chamber (hemacytometer) or Countess automated cell counter.
The 10× Genomics platform recommends 1,000 cells/μl, but you could aim for 700-1,000 cells per μl. In cases where the cell suspension is too diluted, perform an additional spin in a cold benchtop centrifuge and resuspend the cells in a lower volume of PBS containing 0.5% BSA.
Follow the protocol for cell encapsulation, cDNA amplification, and library preparation provided by 10× Genomics.
Alternative protocol:
The Optiprep step can be avoided if the sample preparation is clean with negligible amounts of large tissue clumps or debris, which may accumulate during the bleeding procedure. Below is a quick alternative protocol without the Optiprep step. Importantly, this method may help to obtain a higher number of hemocytes as compared with the Optiprep gradient method.
Follow Steps B1-B8.
After bleeding, transfer the cell suspension to clean RNase-free Eppendorf tubes (not LoBind) by filtering through a 100-μm cell strainer.
Note: Do not use LoBind tubes during the centrifugation steps.
Centrifuge the cell suspension at 1,000 × g for 5 min at 4°C.
Carefully remove the supernatant from the cell pellet and resuspend the cells in 100 μl 0.5% BSA in PBS.
Repeat steps 3-4 for a total of two washes and transfer the final cell suspension to a new Eppendorf LoBind tube.
Note: LoBind tubes are recommended at this final step as they may help to reduce cell loss.
Count the cells using a Neubauer counting chamber (hemacytometer) or Countess automated cell counter. For counting, mix 10 μl 0.4% Trypan Blue solution with 10 μl cell suspension (1:1 dilution) and load onto the counting chamber. Determine the percentage of viable cells, which must be greater than 90%.
Aim for cell numbers recommended by 10× Genomics, which is typically 700-1,000 cells per μl. This is the ‘sweet spot' for achieving a 10,000 cell recovery after encapsulation.
Recipes
Optiprep gradient solution concentration
1.09 g/ml in PBS (Optiprep to PBS ratio - 0.27:0.73). Prepare at least 4 ml Optiprep solution and divide into two 15-ml Falcon tubes (i.e., for two samples). More details about Optiprep can be found at https://www.optiprep.com/C14.pdf.
0.5% BSA in 1× PBS
Add 0.5 g ultrapure BSA to 100 ml 1× PBS. Mix thoroughly and filter using a 0.22-μm sterile filter unit. Scale down the volume depending on the sample size.
20% glycerol solution
Add 200 ml pure glycerol to 800 ml distilled water. Scale down the volume as required.
Acknowledgments
We thank Dr. Stephanie E. Mohr and Dr. Ram Viswanatha for suggestions and input regarding the protocol. N.P is an investigator at Howard Hughes Medical Institute. This protocol has been demonstrated to work for scRNA-seq of Drosophila larval blood (Tattikota et al., 2020). Figure 2 was generated with the help of Biorender (https://app.biorender.com/).
Competing interests
The authors declare no competing interests.
References
- Banerjee, U., Girard, J. R., Goins, L. M. and Spratford, C. M. (2019). Drosophila as a Genetic Model for Hematopoiesis. Genetics 211(2): 367-417.
- Gold, K. S. and Bruckner, K. (2015). Macrophages and cellular immunity in Drosophila melanogaster. Semin Immunol 27(6): 357-368.
- Klein, A. M., Mazutis, L., Akartuna, I., Tallapragada, N., Veres, A., Li, V., Peshkin, L., Weitz, D. A. and Kirschner, M. W. (2015). Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161(5): 1187-1201.
- Macosko, E. Z., Basu, A., Satija, R., Nemesh, J., Shekhar, K., Goldman, M., Tirosh, I., Bialas, A. R., Kamitaki, N., Martersteck, E. M., Trombetta, J. J., Weitz, D. A., Sanes, J. R., Shalek, A. K., Regev, A. and McCarroll, S. A. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161(5): 1202-1214.
- Markus, R., Laurinyecz, B., Kurucz, E., Honti, V., Bajusz, I., Sipos, B., Somogyi, K., Kronhamn, J., Hultmark, D. and Ando, I. (2009). Sessile hemocytes as a hematopoietic compartment in Drosophila melanogaster. Proc Natl Acad Sci U S A 106(12): 4805-4809.
- Petraki, S., Alexander, B. and Bruckner, K. (2015). Assaying Blood Cell Populations of the Drosophila melanogaster Larva. J Vis Exp(105).
- Tattikota, S. G., Cho, B., Liu, Y., Hu, Y., Barrera, V., Steinbaugh, M. J., Yoon, S. H., Comjean, A., Li, F., Dervis, F., Hung, R. J., Nam, J. W., Ho Sui, S., Shim, J. and Perrimon, N. (2020). A single-cell survey of Drosophila blood. Elife 9: e54818.
- Vlisidou, I. and Wood, W. (2015). Drosophila blood cells and their role in immune responses. FEBS J 282(8): 1368-1382.
- Zheng, G. X., Terry, J. M., Belgrader, P., Ryvkin, P., Bent, Z. W., Wilson, R., Ziraldo, S. B., Wheeler, T. D., McDermott, G. P., Zhu, J., Gregory, M. T., Shuga, J., Montesclaros, L., Underwood, J. G., Masquelier, D. A., Nishimura, S. Y., Schnall-Levin, M., Wyatt, P. W., Hindson, C. M., Bharadwaj, R., Wong, A., Ness, K. D., Beppu, L. W., Deeg, H. J., McFarland, C., Loeb, K. R., Valente, W. J., Ericson, N. G., Stevens, E. A., Radich, J. P., Mikkelsen, T. S., Hindson, B. J. and Bielas, J. H. (2017). Massively parallel digital transcriptional profiling of single cells. Nat Commun 8: 14049.
- Zilionis, R., Nainys, J., Veres, A., Savova, V., Zemmour, D., Klein, A. M. and Mazutis, L. (2017). Single-cell barcoding and sequencing using droplet microfluidics. Nat Protoc 12(1): 44-73.
文章信息
版权信息
![]() Tattikota and Perrimon. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
Tattikota and Perrimon. This article is distributed under the terms of the Creative Commons Attribution License (CC BY 4.0).
如何引用
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Tattikota, S. G. and Perrimon, N. (2021). Preparation of Drosophila Larval Blood Cells for Single-cell RNA Sequencing. Bio-protocol 11(16): e4127. DOI: 10.21769/BioProtoc.4127.
- Tattikota, S. G., Cho, B., Liu, Y., Hu, Y., Barrera, V., Steinbaugh, M. J., Yoon, S. H., Comjean, A., Li, F., Dervis, F., Hung, R. J., Nam, J. W., Ho Sui, S., Shim, J. and Perrimon, N. (2020). A single-cell survey of Drosophila blood. Elife 9: e54818.
分类
发育生物学 > 细胞生长和命运决定 > 分化
免疫学 > 免疫细胞分离 > 骨髓细胞
细胞生物学 > 细胞分离和培养 > 细胞分离
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。