

Computational Analysis and Phylogenetic Clustering of SARS-CoV-2 Genomes
SARS-CoV-2基因组计算分析及系统进化聚类分析
发布: 2021年04月20日第11卷第8期 DOI: 10.21769/BioProtoc.3999 浏览次数: 7280
评审: Tharmarajan RamprasathAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Sep 2020
Advertisement










Abstract
COVID-19, the disease caused by the novel SARS-CoV-2 coronavirus, originated as an isolated outbreak in the Hubei province of China but soon created a global pandemic and is now a major threat to healthcare systems worldwide. Following the rapid human-to-human transmission of the infection, institutes around the world have made efforts to generate genome sequence data for the virus. With thousands of genome sequences for SARS-CoV-2 now available in the public domain, it is possible to analyze the sequences and gain a deeper understanding of the disease, its origin, and its epidemiology. Phylogenetic analysis is a potentially powerful tool for tracking the transmission pattern of the virus with a view to aiding identification of potential interventions. Toward this goal, we have created a comprehensive protocol for the analysis and phylogenetic clustering of SARS-CoV-2 genomes using Nextstrain, a powerful open-source tool for the real-time interactive visualization of genome sequencing data. Approaches to focus the phylogenetic clustering analysis on a particular region of interest are detailed in this protocol.
Keywords: COVID-19 (COVID-19)Background
Severe Acute Respiratory Syndrome- related coronaviruses (SARS-CoV) are one of the largest single-stranded RNA virus families known to date (Zhu et al., 2020). Recently, SARS-CoV-2, a novel strain of coronavirus, has been identified as the causal pathogen for the ongoing Coronavirus disease 2019 (COVID-19) pandemic (Huang et al., 2020). The infectious disease that first originated in Wuhan, China, spread to other nations at an alarmingly rapid pace. With 3,517,345 cases reported globally and a death toll of 243,401 (as of 5th May 2020), the disease continues to be a public health concern and a potential threat to the socio-economic welfare of nations and healthcare systems worldwide (World Health Organization, 2020. Novel Coronavirus (2019-nCoV): situation report, 106).
Owing to the rapid advancement of next-generation sequencing (NGS) technology and analysis methods, sequencing the viral genome has been recognized as a viable tool to aid the diagnosis and treatment of COVID-19 and help to understand the disease epidemiology. As the disease evolves over time, more sequencing data for SARS-CoV-2 genomes is being made available in the public domain. To date, there are over 25,000 publicly available genomes of SARS-CoV-2 from different geographical origins. Phylogenetic principles have previously been successfully utilized to contain and diffuse recent pandemic events such as avian influenza, the Zika virus epidemic, and HIV (Salemi et al., 2008; Babakir-Mina et al., 2009; Angeletti et al., 2016). With the rapid accumulation of sequencing data, phylogenetic and phylodynamic analysis are potentially powerful tools for studying the evolutionary patterns of rapidly evolving RNA viruses, and therefore help to understand the epidemiology of the outbreak.
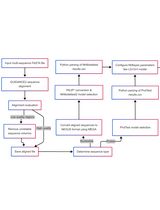
Visualizing evolutionary epidemiology can help to provide a deeper understanding of the global diversity of SARS-CoV-2. Nextstrain is an open-source project that aims to provide real-time interactive visualization of rapidly evolving pathogens coupled with additional data such as geographic information (Hadfield et al., 2018). Nextstrain utilizes Augur, a bioinformatics toolkit for the systematic analysis of genome sequences, and Auspice, an interactive web service for the visualization of analysis results. This protocol has been created to aid bioinformaticians in gaining an epidemiological understanding of the SARS-CoV-2 pathogen using the powerful phylogenetic analysis toolkit provided by Nextstrain. The data and parameters used in this protocol are specific to SARS-CoV-2 genomes; however, Nextstrain is a generalized toolkit for the analysis of pathogen phylogenies and can be customized using the appropriate data and parameters suited to the pathogen of interest. All software and datasets used in this protocol are available in the public domain.
Equipment
We explicitly assume that the user has some experience working with shell commands on a Linux-based operating system and has superuser privileges.
Computational Requirements
We recommend using a workstation or a server with a 64 bit Linux-based operating system, possessing 8 GB RAM and sufficient hard disk space (at least 250 GB) to store the files used and produced in this analysis. The commands given in this analysis protocol have been validated on Ubuntu (18.04 LTS) Linux Distribution.
Software
Required Software
This protocol uses the following tools and Nextstrain software to perform the phylogenetic analysis:
Docker Engine (https://www.docker.com/)
Anaconda (https://www.anaconda.com/)
Nextstrain ( Hadfield et al., 2018 )
Augur ( Hadfield et al., 2018 )
MAFFT (Katoh and Standley, 2013)
IQTREE ( Nguyen et al., 2015 )
All requisite tools and their dependents must be installed before proceeding with the analysis.
Datasets
The protocol uses the SARS-CoV-2 genome sequence datasets made available by the Global Initiative on Sharing All Influenza Data (GISAID) (Shu and McCauley, 2017).
The installation steps for all tools used in this protocol and the instructions for downloading the requisite datasets are given in the following section.
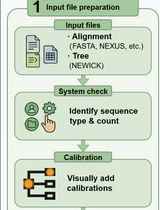
Procedure
文章信息
版权信息
© 2021 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Jolly, B. and Scaria, V. (2021). Computational Analysis and Phylogenetic Clustering of SARS-CoV-2 Genomes. Bio-protocol 11(8): e3999. DOI: 10.21769/BioProtoc.3999.
分类
系统生物学 > 基因组学 > 种系遗传学
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。