

HoSeIn: A Workflow for Integrating Various Homology Search Results from Metagenomic and Metatranscriptomic Sequence Datasets
HoSeIn:整合来自宏基因组和元转录组序列数据集的各种同源性搜索结果的工作流

发布: 2020年07月20日第10卷第14期 DOI: 10.21769/BioProtoc.3679 浏览次数: 4908
评审: Prashanth N SuravajhalaPaola Monica TaliaAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Mar 2013
Advertisement









Abstract
Data generated by metagenomic and metatranscriptomic experiments is both enormous and inherently noisy. When using taxonomy-dependent alignment-based methods to classify and label reads, the first step consists in performing homology searches against sequence databases. To obtain the most information from the samples, nucleotide sequences are usually compared to various databases (nucleotide and protein) using local sequence aligners such as BLASTN and BLASTX. Nevertheless, the analysis and integration of these results can be problematic because the outputs from these searches usually show inconsistencies, which can be notorious when working with RNA-seq. Moreover, and to the best of our knowledge, existing tools do not criss-cross and integrate information from the different homology searches, but provide the results of each analysis separately. We developed the HoSeIn workflow to intersect the information from these homology searches, and then determine the taxonomic and functional profile of the sample using this integrated information. The workflow is based on the assumption that the sequences that correspond to a certain taxon are composed of:
1)sequences that were assigned to the same taxon by both homology searches;
2)sequences that were assigned to that taxon by one of the homology searches but returned no hits in the other one.
Background
The microbiome can be characterised and its potential function inferred using metagenomics, whereas metatranscriptomics provides a snapshot of the active functional (and taxonomic) profile of the microbial community by analysing the collection of expressed RNAs through high-throughput sequencing of the corresponding cDNAs (Marchesi and Ravel, 2015). Data generated by metagenomic and metatranscriptomic experiments is both enormous and inherently noisy (Wooley et al., 2010). The pipelines used to analyse this kind of data normally include three main steps: (1) pre-processing and (2) processing of the reads, and (3) downstream analyses (Aguiar-Pulido et al., 2016). Pre-processing mainly involves removing adapters, filtering by quality and length, and preparing data for subsequent analysis (Aguiar-Pulido et al., 2016). After pre-processing the reads, the next step (processing) consists in classifying each read according to the organism with the highest probability of being the origin of that read. This classification and labelling can be either taxonomy-dependent or independent. Taxonomy-dependent methods use reference databases, and these can be further classified as alignment-based, composition-based, or hybrid. Alignment-based methods usually give the highest accuracy but are limited by the reference database and the alignment parameters used, and are generally computation and memory intensive. Composition-based methods have not yet achieved the accuracy of alignment-based approaches, but require fewer computational resources because they use compact models instead of whole genomes (Aguiar-Pulido et al., 2016). Taxonomy-independent methods do not require a priori knowledge because they separate reads based on certain properties (distance, k-mers, abundance levels, and frequencies) (Aguiar-Pulido et al., 2016).
Once the reads have been classified or labelled as best as possible, downstream analyses (step 3) attempt to extract useful knowledge from the data, such as the potential (metagenomics) or active (metatranscriptomics) functional profile. There are various useful resources for the functional annotation of the genes to which the reads are mapped, such as functional databases–gene ontology (GO) (Ashburner et al., 2000; Blake et al., 2015), Kyoto Encyclopaedia of Genes and Genomes (KEGG) (Ogata et al., 1999; Kotera et al., 2015), Clusters of Orthologous Groups (COG) (Tatusov, 2000), InterPRO (Finn et al., 2017), SPARCLE (Marchler-Bauer et al., 2017), and SEED (Overbeek et al., 2014)–and other tools that can also be used to obtain functional profiles. Among the latter, some are web-based, such as MG-RAST (Glass and Meyer, 2011) and IMG/M (Markowitz et al., 2012), and others are standalone programs, like MEGAN (Huson et al., 2007). MEGAN uses the NCBI taxonomy to classify the results from the homology searches, and uses reference InterPRO (Finn et al., 2017), EggNOG (Powell et al., 2012), KEGG (Ogata et al., 1999) and SEED (Overbeek et al., 2014) databases to perform functional assignment.
The same suite of tools can be used to perform taxonomic assignments of metagenomic and metatranscriptomic data. Nevertheless, in both cases the same limitations are encountered, including algorithms that have to process large volumes of data (short reads), and the paucity of reference sequences in the databases. Additionally, most of these tools only use a subset of available genomes or focus on certain organisms, and many do not include eukaryotes. On the other hand, there are major differences in how each workflow determines the taxonomic profile, because some perform searches against protein databases, whereas others do so in a nucleotide space (a review can be found in Shakya et al., 2019). Our HoSeIn workflow (from Homology Search Integration) centres on the processing and downstream analyses steps, and we developed it for using with taxonomy-dependent alignment-based methods (Video 1). As we already mentioned, the latter use homology searches against sequence databases as the first step to classify and label reads. To obtain as much information as possible from the samples, the nucleotide datasets are compared to nucleotide and protein databases using local sequence aligners such as BLAST (Altschul et al., 1990) or FASTA (Pearson, 2004). Nevertheless, once the homology searches are complete, the analysis and integration of these results can be problematic because the outputs from these searches usually show differences and inconsistencies, which can be particularly notorious when working with RNA-seq (Video 1 and Figure 1). On one hand, amino acid- based searches can detect organisms distantly related to those in the reference database but are prone to false discovery. In contrast, nucleotide searches are more specific but are unable to identify insufficiently conserved sequences. Consequently, taxonomic and functional profiles should be carefully interpreted when they are assigned using one or the other. For example, assignments using searches against nucleotide databases, especially for protein coding genes, are likely to be less effective if no near neighbours exist in the reference databases. In this respect, and to the best of our knowledge, existing tools do not intersect information from the different homology searches to integrate the different results, but provide the results of each analysis separately. We developed the HoSeIn workflow to criss-cross the information from both homology search results (nucleotide and protein) and then perform final assignments on the basis of this integrated information. Sequences are assigned to a certain taxon if they were assigned to that taxon by both homology searches, and if they were assigned to that taxon by one of the homology searches but returned no hits in the other one (Video 1 and Figure 1). Specifically, our workflow extracts all the available information for each sequence from the different tools that were used to process the dataset (homology searches and whatever method was used to classify and label the sequences, for example MEGAN [Huson et al., 2007]), and uses it to build a local database. The data for each sequence is then intersected to define the taxonomic profile of the sample following the above-mentioned criteria. Consequently, the main novelty of our workflow is that final assignments integrate results from both homology searches, capitalising on their strengths and thus making them more robust and reliable (Video 1). For metatranscriptomics in particular, where results are difficult to interpret, this represents a very useful tool.

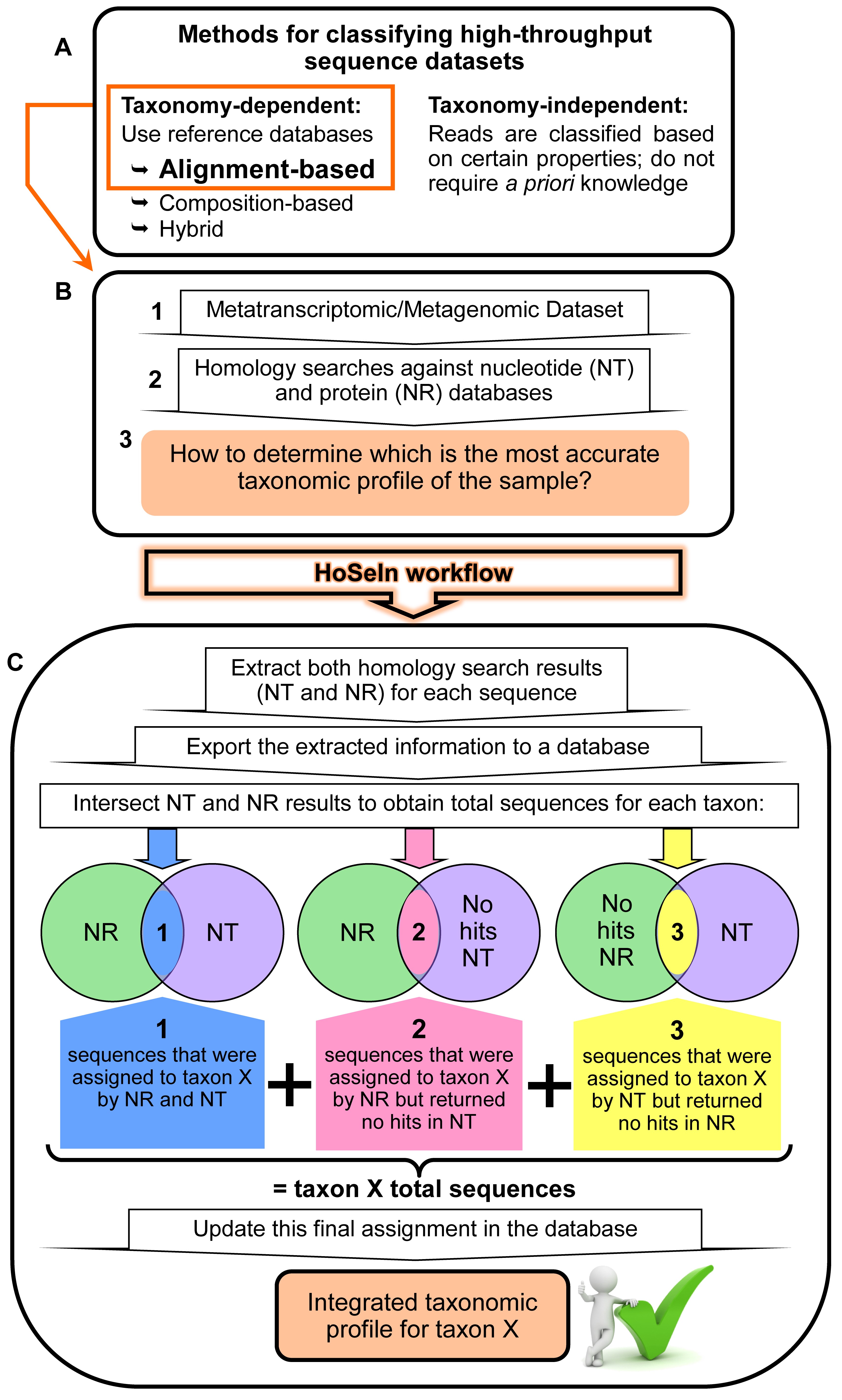
Figure 1. Rationale behind the HoSeIn workflow. A. There are various methods for determining the taxonomic profile of a microbiome in a sequencing-based analysis, and these can be taxonomy dependent or independent (see text for details). B. When using taxonomy-dependent alignment-based methods to analyse metagenomic or metatranscriptomic datasets (1), these are usually compared to nucleotide and protein databases using local sequence aligners such as BLAST (Altschul et al., 1990) or FASTA (Pearson, 2004) (2). Nevertheless, the analysis and integration of these results can be problematic because the outputs from these searches usually show inconsistencies (3). C. The HoSeIn workflow intersects the information from both homology search results and final assignments are determined on the basis of this integrated information. In this way, sequences are assigned to a certain taxon if they were assigned to that taxon by both homology searches (1), and if they were assigned to that taxon by one of the homology searches but returned no hits in the other one (2 and 3).
Equipment
- Desktop computer with an Intel Core i7 2600 processor (3,40 Ghz, 8 Mb, 4 Cores, 8 Threads, video and Turboboost); Intel DH67BL Motherboard, LGA 1155 socket; with 7.1 + 2 sound; 1 Gb network; RAID 0,1,5 y 10; and four Kingston 1.333 Mhz DDR3 4 GB memories
Software
- Ubuntu 18.04.3 LTS (Ubuntu, https://ubuntu.com/#download), last accessed on 11/9/2019
- BLAST (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/), blast-2.2.25+ last accessed 2/7/2013
Note: FASTA programs (FASTA DNA:DNA and FASTX) can also be used for the homology searches. Nevertheless, BLAST and FASTA programs represent a major computational bottleneck when aligning high-throughput datasets against protein databases, and different tools have recently been developed to improve performance. In particular, DIAMOND is an open-source sequence aligner for protein and translated DNA searches which performs at 500x-20,000x the speed of BLAST, is suitable for running on standard desktops and laptops, and offers various output formats as well as taxonomic classification (Buchfink et al., 2015). Thus, when aligning large datasets against protein databases with limited computational resources, we recommend using Diamond. - MEGAN6 (http://ab.inf.uni-tuebingen.de/software/megan6/); MEGAN_Community_windows-x64_6_17_0 version last accessed on 18/9/2019
In this tutorial MEGAN is used to process the homology search output files and then extract the taxonomic and functional information. For downloading and installing this software:- Go to the MEGAN website (http://ab.inf.uni-tuebingen.de/software/megan6) and download the MEGAN6 version that matches your Operating System, as well as the corresponding mapping files
- Run the installer
- Go to the MEGAN website (http://ab.inf.uni-tuebingen.de/software/megan6) and download the MEGAN6 version that matches your Operating System, as well as the corresponding mapping files
- DB Browser for SQLite (DB4S) (https://sqlitebrowser.org/); DB.Browser.for.SQLite-3.11.2-win64 version last accessed on 5/9/2019
DB Browser for SQLite (DB4S) is a high quality, visual, open source tool used to create, design, and edit database files compatible with SQLite. It uses a familiar spreadsheet-like interface, and does not require learning complicated SQL commands. In our workflow we use DB4S to create a local database that includes all the available information for each sequence from the dataset. All this data is then used to define the taxonomic and functional profile of the sample. For downloading and installing this software:- Download the DB4S version that matches your Operating System from the website (https://sqlitebrowser.org/)
- Run the installer
- Download the DB4S version that matches your Operating System from the website (https://sqlitebrowser.org/)
Procedure
文章信息
版权信息
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Rozadilla, G., Moreiras Clemente, J. and McCarthy, C. B. (2020). HoSeIn: A Workflow for Integrating Various Homology Search Results from Metagenomic and Metatranscriptomic Sequence Datasets. Bio-protocol 10(14): e3679. DOI: 10.21769/BioProtoc.3679.
分类
系统生物学 > 基因组学 > 种系遗传学
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。