

Super-resolution Microscopy-based Bimolecular Fluorescence Complementation to Study Protein Complex Assembly and Co-localization
基于高分辨率显微镜的双分子荧光互补用于蛋白复合体装配和共定位

(*contributed equally to this work) 发布: 2020年02月20日第10卷第4期 DOI: 10.21769/BioProtoc.3524 浏览次数: 6339
评审: David PaulSijie WeiYosuke Senju
参见作者原研究论文
The authors used this protocol in:

May 2019
Advertisement









Abstract
Numerous experimental approaches exist to study interactions between two subunits of a large macromolecular complex. However, most methods do not provide spatial and temporal information about binding, which are critical for dissecting the mechanism of assembly of nanosized complexes in vivo. While recent advances in super-resolution microscopy techniques have provided insights into biological structures beyond the diffraction limit, most require extensive expertise and/or special sample preparation, and it is a challenge to extend beyond binary, two color experiments. Using HyVolution, a super-resolution technique that combines confocal microscopy at sub-airy unit pinhole sizes with computational deconvolution, we achieved 140 nm resolution in both live and fixed samples with three colors, including two fluorescent proteins (mTurquoise2 and GFP) with significant spectral overlap that were distinguished by means of shifting the excitation wavelength away from common wavelengths. By combining HyVolution super-resolution fluorescence microscopy with bimolecular fluorescence complementation (SRM-BiFC), we describe a new assay capable of visualizing protein-protein interactions in vivo at sub-diffraction resolution. This method was used to improve our understanding of the ordered assembly of the Saccharomyces cerevisiae spindle pole body (SPB), a ~1 giga-Dalton heteromeric protein complex formed from 18 structural components present in multiple copies. We propose that SRM-BiFC is a powerful tool for examination of direct interactions between protein complex subunits at sub-diffraction resolution in live cells.
Keywords: Protein complex assembly (蛋白复合体装配)Background
Protein complexes execute fundamental functions involved in various biological processes. To have these molecular machines assembled in a timely manner in a precise cellular location is critical for cellular activities, including protein synthesis, chromosome segregation, nuclear transport, signaling, motility and other diverse functions. Typically, the formation of many multimeric complexes is thought to occur in a stepwise manner via recruitment and integration of subunits or subcomplexes, which are bridged to existing molecules within the larger assembly by a physical protein-protein contact. Understanding the assembly of large macromolecular complexes is historically the job of biochemists and crystallographers who employ an arsenal of techniques: purification of complexes to determine the identity of subunits, co-purification of proteins, yeast two-hybrid screening, in vitro binding assays, chemical cross-linking studies and structural analysis to understand protein-protein interactions, and finally, reconstitution of the complex to determine the hierarchy of assembly.
While this traditional approach has been successful in many cases, a major challenge is incorporating knowledge from studies on a handful of proteins to the larger and more relevant problem of macromolecular complex assembly within the cell. In cells, the timing of protein production, protein localization and post-translational modifications affect distribution, local concentrations and interactors that in turn influence complex assembly. A number of imaging-based techniques have been developed to study protein-protein interactions within cells, including fluorescent resonance energy transfer (FRET), which examines energy transfer between pairwise donor-acceptor fluorophores linked to proteins of interest; fluorescence cross-correlation spectroscopy (FCCS), which measures the diffusion pattern of two fluorescent molecules through the focal volume; and bimolecular fluorescence complementational (BiFC), which is based on ability of proteins in close proximity to self-complement and create a functional fluorescent protein from two non-fluorescent protein fragments (Bacia et al., 2006; Slaughter et al., 2007; Kerppola, 2008; Miller et al., 2015; Unruh et al., 2018). These techniques can be used in live cells to study endogenously expressed protein-protein interactions in a pairwise manner provided the molecules of interest can be fused to fluorescent proteins (FP). In yeast, genes can be easily tagged at the N- or C-termini using PCR-based methods (Gardner and Jaspersen, 2014). With the development of CRISPR/Cas9 methodologies, genome editing is becoming increasingly feasible such that these approaches can be applied in a wide array of systems. The use of endogenous proteins, rather than transgenes, greatly minimizes false positives caused by high levels of protein expression. In comparison to in vitro assays, FRET, FCCS and BiFC have the advantage of indicating where within a cell the protein-protein interaction occurs (for example, see Muller et al., 2005; Cabantous and Waldo, 2006; Kerppola, 2006; Slaughter and Li, 2010; Sung et al., 2013; Chen et al., 2014; Smoyer et al., 2016; Hennen et al., 2018). However, these methods are limited in their spatial resolution due to the diffraction limit (~200 nm for most FPs), and combining FRET or FCCS with super-resolution methods in cells is extremely challenging.
Structured illumination microscopy (SIM), stimulated emission depletion (STED), stochastic optical reconstruction microscopy (STORM) and photoactivated localization microscopy (PALM) are examples of super-resolution microscopy (SRM) approaches that are pushing cell biology in new directions once thought impossible by beating the diffraction limit (Huang et al., 2009; Schermelleh et al., 2010). The ~10-100 nm resolution achievable by these methods has enhanced our overall understanding of cell biology, particularly at the level of organelles, membranes and other cellular structures. The application of SRM to protein-protein interactions within nanosized protein complexes has been slower to develop, in part because it is difficult to study endogenous proteins with many of these methods, particularly if multiple fluorophores/FPs are needed to monitor interactions and to provide orientation of where within the complex or the assembly cycle the interaction is occurring. While a plethora of FPs have been developed, linear unmixing to image multiple fluorophores has not been integrated into SRM yet, limiting the use of many combinations of FPs. To overcome this obstacle, we combined a version of BiFC based on split-GFP to assay protein-protein interactions (Figure 1).To provide spatial and temporal information as to where and when binding within our complex of interest occurs at sub-diffraction resolution, we developed a three-color imaging strategy using HyVolution with 140 nm lateral resolution (Chen et al., 2019). Importantly, our protocol allows for imaging of endogenously expressed proteins in live cells fused to mTurquoise2 and GFP, along with mCherry, without spectral cross-talk. While the method was developed in Saccharomyces cerevisiae, it is suitable for a wide variety of systems, and it greatly expands our ability to inspect the step-wise assembly pathway of heteromeric macromolecular machines by elucidating protein-protein interactions in vivo at the nanoscale level.
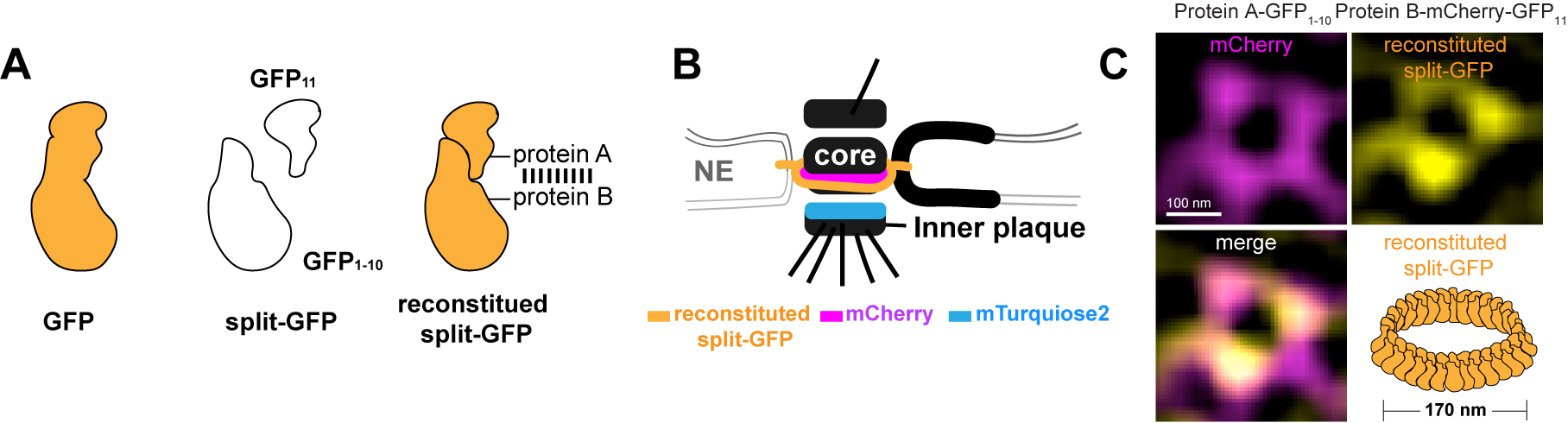
Figure 1. Structured illumination microscopy (SIM)-based split-GFP system to assay protein-protein interaction within nanosized protein complexes. A. Schematic of the bimolecular fluorescence complementation split-GFP system. Two parts of split-GFP can reconstruct and lead to GFP fluorescence when they associate with each other by interaction between their fusion proteins. B. An illustration of the yeast centrosome viewed from the side to show how it is embedded and anchored in the nuclear envelope (NE) by a series of integral membrane proteins, visualized by reconstituted split-GFP and mCherry tagging. The soluble inner-core plaque linker protein was fused to mTurquoise2. C. We showed that our spindle pole body (SPB) protein (fused to GFP1-10) is a component of the membrane complex. In a top down view of the cylinder-shaped SPB, we observed a 170 ring of reconstituted GFP when combined with a known membrane component (fused to mCherry-GFP11) using SIM-BiFC (Chen et al., 2019).
Materials and Reagents
- 0.2 µm filtration unit-150 ml (Thermo Fisher Scientific, catalog number: 150-0020)
- 13 x 100 mm glass culture tube (Fisher, catalog number:14-961-27)
- 15 cm plain wood applicators (Fisher, catalog number: 23-400-102)
- 1.7 ml microcentrifuge tube (VWR, catalog number: 87003-294)
- 25 x 75 mm glass microscope slides (Fisher, catalog number: 12-544-4)
- 22 x 30 mm Number 1.5 coverslips (VWR, catalog number: 48393-150)
- 25-slide humidified chamber (Daigger Scientific, catalog number: EF15989A)
- Yeast strains (see Note 1)
- SLJ10800 (MATa/α SPC42-mTurquiose2-NATMX/Spc42-YFP-HIS3MX bar1/bar1 ADE2/ADE2 trp1-1/TRP1 lys2Δ/LYS2 leu2-3,112/leu2-3,112 his3-11,15/his3-11,15 ura3-1/ura3-1)
- SLJ6209 (MATa SPC42-mCherry-URA3MX bar1 ADE2 trp1-1 lys2Δ leu2-3,112 his3-11,15 ura3-1)
- SLJ7904 (MATa/α SPC110-GFP-KANMX/Spc110 bar1/bar1 ADE2/ade2 trp1-1/TRP1 lys2Δ/LYS2 leu2-3,112/leu2-3,112 his3-11,15/his3-11,15 ura3-1/ura3-1)
- SLJ12889 (MATa Spc110-mTurquiose2-HIS3MX bar1 ADE2 TRP1 lys2Δ leu2-3,112 his3-11,15 ura3-1)
- Bacto-yeast extract
- Bacto-peptone
- Glucose
- Bacto-agar
- Yeast-nitrogen base with ammonium sulfate without amino acids
- CSM powder (Sunrise Science, catalog number: 1001-100)
- 16% paraformaldehyde (Ted Pella, catalog number: 18505)
- Sucrose
- Sodium chloride (NaCl)
- Potassium chloride (KCl)
- Disodium phosphate
- Monopotassium phosphate
- ProLong Diamond mounting medium (Thermo Fisher, catalog number: P36961)
- DeltaVision Immersion Oil Kit; contains bottles of 18 oils with refractive indices from 1.500 to 1.534 (GE Healthcare, catalog number: 29163068)
- Ethanol (Amresco, catalog number: 64-17-5)
- YPD plate (see Recipes)
- SC complete broth (see Recipes)
Note: We typically do not culture cells in YPD liquid media since it produces high background levels of fluorescence. - 4% paraformaldehyde solution (see Recipes)
- PBS (see Recipes)
Equipment
- 2 L flask
- Rotating tube mixer (Fisher Scientific, catalog number: 346)
- 23 °C incubator (VWR, catalog number: 7774)
- Culture roller (New Brunswick TC-7, catalog number: M1053-4004)
- Desktop centrifuge (Eppendorf 5424, catalog number: 022620401)
- Pipettes (Gilson P10, catalog number: F144802)
- BioPhotometer (Eppendorf, model: 6131, catalog number: 62111-460)
- GE Healthcare OMX structured illumination microscope (GE Healthcare DeltaVision OMX Blaze V4) with three scientific complementary metal-oxide semiconductor (sCMOS) cameras and 6 laser lines, or a similar microscope (e.g., Nikon N-SIM, Zeiss Elyria)
- Olympus PLANAPO N 60x, 1.42 NA oil immersion objective
- DeltaVision OMX channel/image registration slide (GE Healthcare, catalog number: 53-852891-000)
- Leica Inverted Confocal SP8 equipped with:
- White Light Laser (470-670 nm range)
- An Argon Laser
- 3 Leica HyD Detectors
- Leica Application Suite software with Hyvolution Module
- Leica 100x, 1.4-NA HC PL APO OIL CS2 Objective
Software
- OMX acquisition computer and software v3.70 (GE Healthcare, http://incelldownload.gehealthcare.com/bin/download_data/OMX/V3.70/DeltaVisionOMX.htm)
- OMX image-processing station with softWoRx v6.52 (GE Healthcare, http://incelldownload.gehealthcare.com/bin/download_data/SoftWoRx/6.5.2/SoftWoRx.htm) software installed
- ImageJ (Java software for image-processing analysis; freely available at http://rsbweb.nih.gov/ij/ or http://Fiji.sc) (Schindelin et al., 2012)
- ImageJ plugins (created in the microscopy center of The Stowers Institute for Medical Research; freely available at http://research.stowers.org/imagejplugins/index.html)
- Leica Application Suite X (LAS X) software for image acquisition (version 3.1.5.16308) (Leica Microsystems)
- Huygens Professional version 17.10.0p5.64b, Scientific Volume Imaging BV, Hilversum, The Netherlands
Procedure
文章信息
版权信息
© 2020 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Readers should cite both the Bio-protocol article and the original research article where this protocol was used:
- Chen, J., Yu, Z., Unruh, J. R., Slaughter, B. D. and Jaspersen, S. L. (2020). Super-resolution Microscopy-based Bimolecular Fluorescence Complementation to Study Protein Complex Assembly and Co-localization. Bio-protocol 10(4): e3524. DOI: 10.21769/BioProtoc.3524.
- Chen, J., Gardner, J. M., Yu, Z., Smith, S. E., McKinney, S., Slaughter, B. D., Unruh, J. R. and Jaspersen, S. L. (2019). Yeast centrosome components form a noncanonical LINC complex at the nuclear envelope insertion site. J Cell Biol 218(5): 1478-1490.
分类
分子生物学 > 蛋白质 > 蛋白质-蛋白质相互作用
细胞生物学 > 基于细胞的分析方法 > 蛋白互作
生物化学 > 蛋白质 > 荧光
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。