

Quantifying Symmetrically Methylated H4R3 on the Kaposi’s Sarcoma-associated Herpesvirus (KSHV) Genome by ChIP-Seq
采用ChIP-Seq定量卡波西肉瘤相关疱疹病毒(KSHV)基因组上的对称甲基化H4R3

发布: 2018年03月20日第8卷第6期 DOI: 10.21769/BioProtoc.2781 浏览次数: 8136
评审: Alka MehraStefan de VriesPrashanth N Suravajhala
参见作者原研究论文
The authors used this protocol in:

Jul 2017







Abstract
Post-translational modifications to histone tails contribute to the three-dimensional structure of chromatin and play an important role in determining the relative expression of nearby genes. One such modification is symmetric di-methylation of arginine residues, which may exhibit different effects on gene expression including blocking the binding of transcriptional activators, or recruiting repressive effector molecules. Recent ChIP-Seq studies have demonstrated the importance of cross-talk between different histone modifications in gene regulation. Thus, to acquire a comprehensive understanding of the combined efforts of these epigenetic marks, ChIP-Seq must be utilized for identifying specific enrichment on the chromatin. Tumorigenic herpesvirus KSHV, employs epigenetic mechanisms for gene regulation, and by evaluating relative abundance of multiple histone modifications in a thorough, unbiased way, using ChIP-Seq, we can get a superior insight concerning the complex mechanisms of viral replication and pathogenesis.
Keywords: KSHV (KSHV)Background
Kaposi’s sarcoma-associated herpesvirus (KSHV) is an oncogenic human virus with two distinct phases during its lifecycle. After initial infection, KSHV establishes a persistent life-long infection in the host that is particularly problematic to the immune-compromised individuals. KSHV can cause various tumors in HIV/AIDS patients including Kaposi’s sarcoma, and multiple B-cell lymphomas (Chang et al., 1994; Cesarman et al., 1995; Russo et al., 1996; Soulier et al., 1995). With a large genome of about 165,000 bp, which encodes nearly 90 different open reading frames, KSHV has ample tools to evade the host immune surveillance system, alter host-cell growth pathways and produce infectious progeny virions.
During the latent phase only a fraction of the viral genes is expressed, which are oncogenic in nature and also help in replication and passaging of the viral episomes into the divided tumor cells (Uppal et al., 2014; Purushothaman et al., 2016). However, many factors including viral co-infection (HIV) and other stimulus such as, hypoxia, oxidative stress, or immune-suppressant medications can trigger the virus to shift into the active, lytic phase of the lifecycle leading to the production of infectious progeny virions, which egress from host cells surface to infect surrounding tissues (Purushothaman et al., 2015). These complex processes are carried out in a coordinated fashion with specific genes expressed in a sequential manner during the switch to a lytic (virus-producing) phase of the viral life cycle (Purushothaman et al., 2015; Aneja and Yuan, 2017).
KSHV employs epigenetic mechanisms to carefully regulate differential gene expression needed to maintain the virus in a specific phase of the lifecycle. Upon infection and entry into the human cells, viral genome acquires cellular histones, similar to the host genomes, and persists in euchromatin (transcription-permissive) or heterochromatin (transcription-repressive) states (Toth et al., 2013; Uppal et al., 2015). By these means, KSHV is able to restrict gene expression during latency to only a minimal subset of genes and is yet poised to rapidly shift into lytic replication phase. The development of KSHV-induced malignancies involves both phases of the lifecycle, thus it is essential for researchers to have a clear understanding of the mechanisms of how these epigenetic changes regulate viral genes expression.
Epigenetic regulation of gene expression relies on conformational changes to chromatin that alter the availability of certain proteins for transcription (Bernstein et al., 2007). One of the best-studied ways organisms induce conformational changes to the chromatin is through histone-tail modifications. Histone residues may be ‘modified’ by the addition of different molecules to specific residues on the histone proteins (Bannister and Kouzarides, 2011). While several modifications have been identified, two of the most-commonly studied types are lysine residue acetylation or lysine residue methylation and they can dictate the transcriptional state of the genes occupied by those modified histones (Zhang et al., 2015). For example, lysine acetylation is generally considered an ‘activating’ mark that upregulates gene expression. Another activating mark is histone 3, lysine 4 trimethylation (H3K4me3), yet another modification on histone H3 at lysine 27 in the same fashion (H3K27me3) is a ‘repressive’ mark, leads to transcriptional silencing (Bannister and Kouzarides, 2011).
Histone lysine acetylation (H3ac), H3K4me3, and H3K27me3 levels have been assessed on the KSHV genome but the landscape of other histone modifications during lytic reactivation had not yet been ascertained (Gunther and Grundhoff, 2010; Toth et al., 2010 and 2013). So, when proteomic interaction studies conducted in our lab suggested that a lytic viral protein, ORF59, could be involved in chromatin remodeling, particularly in regards to arginine methylation, we set out to study the histone arginine methylation. Our recent study, ‘KSHV encoded ORF59 modulates histone arginine methylation of the viral genome to promote viral reactivation’ examined the enrichment of a specific histone modification H4R3me2s (histone 4 arginine 3 symmetric di-methylation) across the viral genome (Strahan et al., 2017). H4R3me involves first the mono-methylation of the arginine followed by the addition of another methyl group in either a symmetric, or asymmetric fashion (H4R3me2s or H4R3me2a, respectively) (Di Lorenzo and Bedford, 2011). Arginine methylation is important for various several cellular processes including RNA processing, DNA repair, transcription, signal transduction, and chromatin remodeling (Pahlich et al., 2006). While the addition of methyl groups on arginine residues increases hydrophobicity that blocks hydrogen bonding but the overall charge is not altered so the binding between nucleic acids or other proteins remains undisturbed (Pahlich et al., 2006). Various protein arginine methyltransferases (PRMTs) are expressed in multiple subcellular locales to modify the protein arginine residues of nuclear as well as cytoplasmic proteins (Bedford and Clarke, 2009).
Interestingly, the conformational difference between asymmetrically modified H4R3me2 and symmetrically H4R3me2 affects transcriptional activation/repression very distinctly. Symmetrically modified H4R3me2 favors the repression of gene expression and transcriptional silencing, while in contrast asymmetrically modified H4R3me2 favors upregulation of gene transcription (Di Lorenzo and Bedford, 2011).
In order to specifically capture the symmetrically modified, H4R3me2s chromatin, it was first absolutely essential to verify that the antibody used for ChIP-Seq purposes would not cross-react with H4R3me2a. Anti-H4R3me2s antibody was obtained and the specificity to the symmetrically modified H4R3me2 was tested before using for immunoprecipitating chromatin from KSHV infected cells. Following the confirmation of its specificity, we proceeded to perform ChIP-Seq from latent and lytically reactivating KSHV positive TRExBCBL1-RTA cells. The advantage of using TRExBCBL1-RTA cells (KSHV infected cell line) was to induce the expression of Replication and Transcription Activator (RTA) by tetracycline/doxycycline, which is both necessary and sufficient to trigger lytic reactivation (Nakamura et al., 2003). The ChIP assay was performed similarly to previously done ChIP-Seq assays that include the following basic steps: harvest the cells and cross-link with formaldehyde, isolate and shear chromatin, immunoprecipitate DNA-protein complexes of interest, purify the DNA, and prepare sequencing libraries from the immunoprecipitated and respective inputs DNA (Figure 1). To obtain a more thorough understanding of the chromatin remodeling role of viral protein ORF59, we needed to test the relative enrichment of several different factors at the KSHV genome including H4R3me2s, ORF59, PRMT5, COPR5. Each of these ChIPs were done in triplicate and samples were combined for library preparation. ChIP-Seq experiments traditionally use approximately 20 million cells per sample; however, to quantify H4R3me2s (and enrichment of other factors as well) on the KSHV genome, we chose to use a modified Low-Cell ChIP protocol with 5 million cells per sample instead (Park, 2009). To accomplish this, the chromatin-shearing step of the ChIP assay was optimized using the Diagenode Bioruptor® Pico to improve the efficiency of the DNA immunoprecipitation. 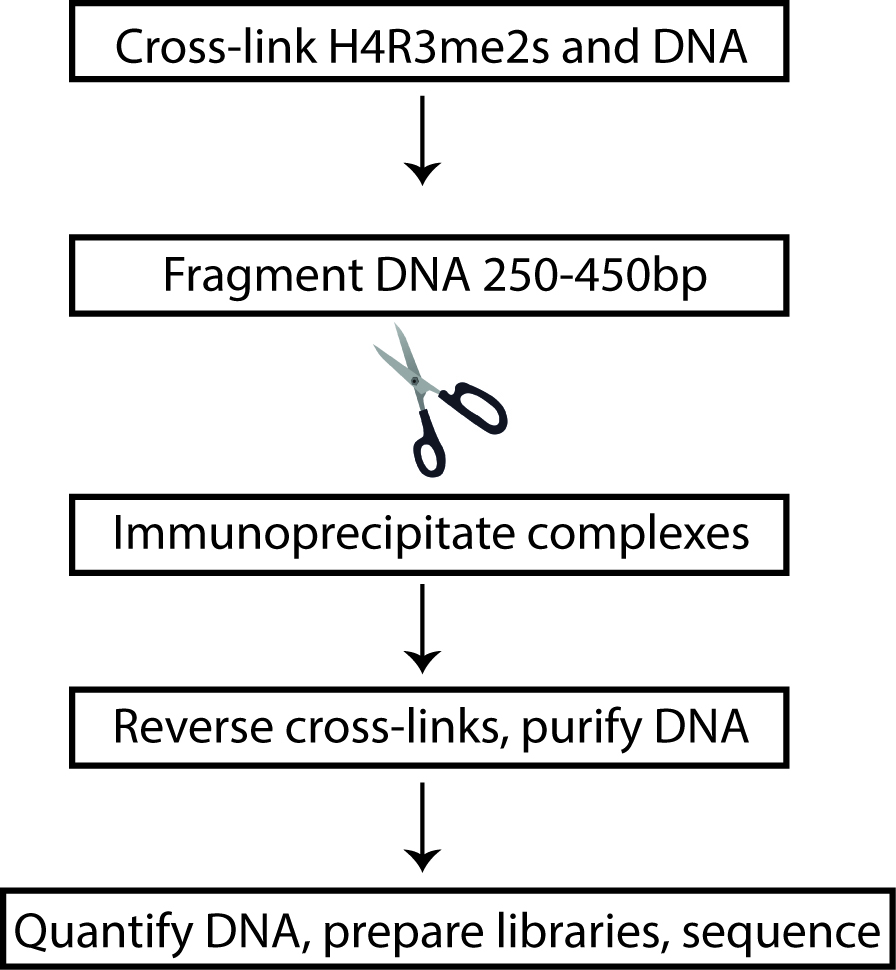
Figure 1. Flow-chart depiction of H4R3me2s ChIP. Cells from latent and lytic KSHV-positive cells were cross-linked to preserve DNA-protein interactions and the DNA was sheared into small fragments. DNA fragments were then immunoprecipitated, purified, and subjected to next-generation sequencing.
Another unique challenge faced in assessing chromatin structure of viral genomes is that H4R3me2s ChIP assay isolates both viral and cellular host DNA bound to H4R3me2s; and furthermore, upon lytic reactivation viral genomes are multiplied resulting in drastically different levels of viral DNA between two samples with an approximately identical number of cells (Figure 2). For this reason, we assessed H4R3me2s levels at a very early time point during lytic reactivation (12 h), before viral genome copies have had a chance to accumulate and possibly bias the downstream ChIP-Seq. As a result of these careful adjustments, we were able to successfully quantify relative enrichment of a repressive chromatin mark on the viral genome during two different phases of the lifecycle and demonstrate a novel chromatin-remodeling role for the early viral protein, ORF59.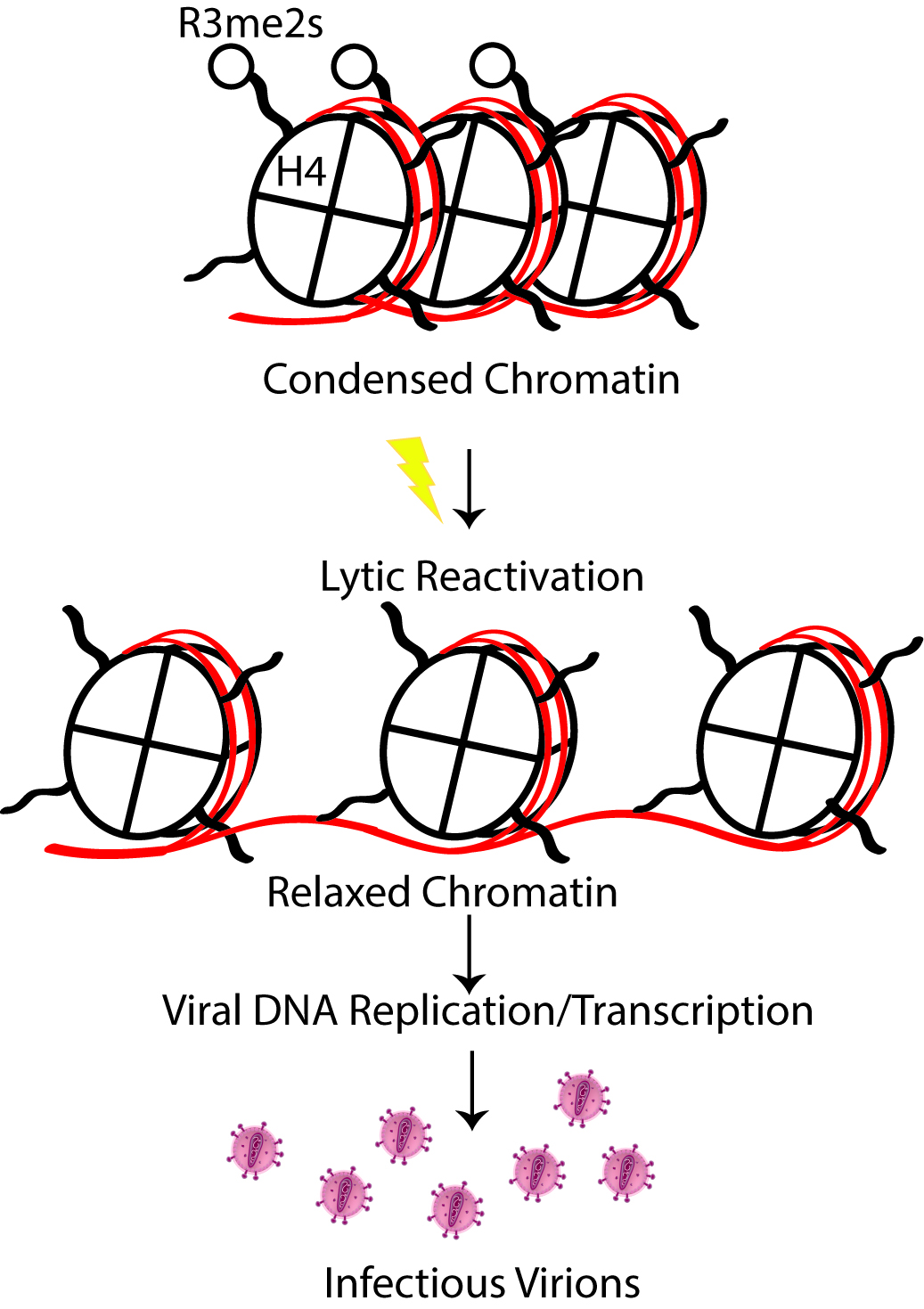
Figure 2. Schematic depiction of H4R3me2s role in KSHV lytic reactivation. H4R3me2s is an abundant hallmark of transcriptionally silent, heterochromatin and must be removed to favor active gene transcription.
Materials and Reagents
- Materials
- Pipette tips
- 1.5 ml Bioruptor® Pico Microtubes with Caps (Diagenode, catalog number: C30010016 )
- Illumina NextSeq 500 Mid Output KT v2 (150 cycles) (Illumina, catalog number: FC-404-2001 )
Note: Researchers should select the most appropriate flow cell for their experimental specifications. To sequence arginine methylation on the KSHV genome, this specific flow cell was sufficient.
- Pipette tips
- Chemicals/stock solutions
- Protease inhibitors (leupeptin, aprotinin, sodium fluoride, pepstatin, and phenylmethylsulfonyl fluoride) (Sigma-Aldrich, catalog number: S8830 )
- Pierce 16% formaldehyde (w/v), Methanol-Free (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: 28906 )
- 1 M glycine
- Bioruptor® Pico sonication beads (Diagenode, catalog number: C01020031 )
- RNase A, 20 mg/ml
- 1.5% agarose gel
- Specific ChIP grade antibodies
Note: To quantify H4R3me2s on the KSHV genome, following ChIP grade antibodies were used, rabbit anti-H4R3me2s (Active Motif, catalog number: 61187 ), rabbit anti-Histone H4 (Active Motif, catalog number: 61299 ), rabbit anti-control IgG (ChIP grade—Cell Signaling Technology, catalog number: 2729 ). - 1x TE
- 5 M NaCl
- 7.5 M ammonium acetate
- 100% ethanol
- GlycoBlue Coprecipitant (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9516 )
- Proteinase K
- QIAGEN MinElute PCR purification Kit (QIAGEN, catalog number: 28004 )
- Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific, InvitrogenTM, catalog number: Q32851 )
- NEXTflexTM ChIP Seq Kit (Illumina compatible) (Bioo Scientific, catalog number: NOVA-5143-02 )
- NEXTflexTM ChIP Seq Barcodes - 12 (Illumina compatible) (Bioo Scientific, catalog number: NOVA-514120 )
- KAPA Library Quantification Complete Kit (Universal), kit code KK4824 (Kapa Biosystems, catalog number: 07960140001 )
- Agilent Bioanalyzer High Sensitivity DNA chip Kit (Agilent Technologies, catalog number: 5067-4626 )
- 0.5 M PIPES, pH 8.0
- 1.7 M KCl
- 10% Nonidet-P40 (NP-40)
- 0.5 M EDTA, pH 8.0
- 1 M Tris-HCl pH 8.1
- Magnetic Protein A, and G beads
- Salmon sperm DNA, sheared 10 mg/ml (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM9680 )
- 10% Triton X-100
- 10% SDS
- 1 M NaHCO3
- Protease inhibitors (leupeptin, aprotinin, sodium fluoride, pepstatin, and phenylmethylsulfonyl fluoride) (Sigma-Aldrich, catalog number: S8830 )
- Buffers
- 1x PBS (137 mM NaCl, 10 mM phosphate, 2.7 mM KCl, and a pH of 7.4)
- Buffer D Chromatin Shearing Buffer (Diagenode, catalog number: C01020030 )
- Cell lysis buffer (see Recipes)
- ChIP dilution buffer (see Recipes)
- ChIP Magnetic A+G beads (see Recipes)
- ChIP Low Salt Wash (see Recipes)
- ChIP elution buffer (see Recipes)
- 1x PBS (137 mM NaCl, 10 mM phosphate, 2.7 mM KCl, and a pH of 7.4)
Equipment
- Pipettes
- Tabletop Eppendorf centrifuge, refrigerated and non-refrigerated
- Bioruptor® Pico Sonication device (Diagenode, catalog number: B01060001 )
- Magnetic stand
- Water bath
- Tube rotators
- Thermocycler
- Qubit Fluorometer (Thermo Fisher Scientific)
- Agilent 2100 Bioanalyzer (Agilent Technologies, model: Agilent 2100 , catalog number: G2939BA)
- Quantitative PCR machine
- Vortexer
- Illumina NextSeq 500 (Illumina, model: NextSeq 500 )
Note: The Illumina NextSeq machine used for these studies is the property of the Nevada Genomics Center, who performed all sequencing runs for this study.
Software
- CLC workbench 10.0.1 (Licensed from QIAGEN, Germantown, MD)
Procedure
文章信息
版权信息
© 2018 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Strahan, R. C., Hiura, K. S. and Verma, S. C. (2018). Quantifying Symmetrically Methylated H4R3 on the Kaposi’s Sarcoma-associated Herpesvirus (KSHV) Genome by ChIP-Seq. Bio-protocol 8(6): e2781. DOI: 10.21769/BioProtoc.2781.
分类
系统生物学 > 表观基因组学 > 测序 > ChIP-seq
微生物学 > 微生物-宿主相互作用 > 病毒
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。