

Assaying the Effects of Splice Site Variants by Exon Trapping in a Mammalian Cell Line
通过外显子捕获检测哺乳动物细胞系中剪接位点变体的作用

发布: 2017年05月20日第7卷第10期 DOI: 10.21769/BioProtoc.2281 浏览次数: 13775
评审: Jia LiAlberto RissoneAnonymous reviewer(s)
参见作者原研究论文
The authors used this protocol in:

Jul 2016
Advertisement









Abstract
There are several in silico programs that endeavor to predict the functional impact of an individual’s sequence variation at splice donor/acceptor sites, but experimental confirmation is problematic without a source of RNA from the individual that carries the variant. With the aid of an exon trapping vector, such as pSPL3, an investigator can test whether a splice site sequence change leads to altered RNA splicing, through expression of reference and variant mini-genes in mammalian cells and analysis of the resultant RNA products.
Keywords: Splicing (剪接)Background
We wished to experimentally test the functional impact of two splice donor site variants, c.760+2T>C and c.3300+2delT, identified in the TEK gene (Souma et al., 2016). As is often the case, samples of cells or mRNA were not available from the individuals carrying these sequence variants, so we utilized the exon trapping method to serve as a functional test. DNA samples were available from patients for PCR amplification of the genomic regions of interest. If patient gDNA samples are unavailable, sequence variants can also be incorporated into wild-type sequence by methods such as PCR-based site-directed mutagenesis.
The exon trapping approach was originally developed to identify unknown exons within long stretches of genomic DNA (Duyk et al., 1990). The pSPL3 exon trapping vector was created to increase the efficiency and reliability of exon identification, and also allowed larger genomic fragments to be screened (Church et al., 1994; Nisson et al., 1994). The pSPL3 vector contains a small artificial gene composed of an SV40 promoter, an exon-intron-exon sequence with functional splice donor and acceptor sites, and a late polyadenylation signal. Within the single intron a multiple cloning site is located, into which a genomic fragment of interest is inserted to create a mini-gene expression construct.
In our example, patient and control genomic DNA fragments from the TEK gene were PCR amplified and cloned between pSPL3 vector exons V1 and V2 using XhoI and BamHI restriction sites. COS-7 cells were then transfected with the mini-gene constructs and the resulting RNA content purified. mRNA transcripts were then reverse transcribed into cDNA. Using vector exon-specific primers, cDNAs produced from the mini-gene constructs were specifically PCR amplified and Sanger sequenced. For the first splice site variant, c.760+2T>C within the 5’ splice site of exon 5, a 1,457 bp genomic fragment of the TEK gene encompassing all of intron 4, exon 5, intron 5, exon 6 and intron 6 was inserted into the construct (Figure 1A). RT-PCR and Sanger sequencing of the mini-gene expressed transcripts showed that the mutation destroyed the splice donor site, which resulted in partial intron 5 inclusion before a cryptic splice site was utilized (Figure 1C). This splicing error is predicted to result in a translational frameshift and premature termination signal, which would likely lead to transcript elimination via the nonsense-mediated decay pathway. For the second splice site variant, c.3300+2delT within the 5’ splice site of exon 22, an 831 bp genomic fragment of the TEK gene encompassing all of intron 21, exon 22 and intron 22 was inserted into the construct (Figure 1B). RT-PCR and Sanger sequencing of the mini-gene expressed transcripts revealed that the splice donor mutation led to skipping of exon 22, which is also predicted to result in a translational frameshift and premature termination signal in the genomic context of the patient (Figure 1C).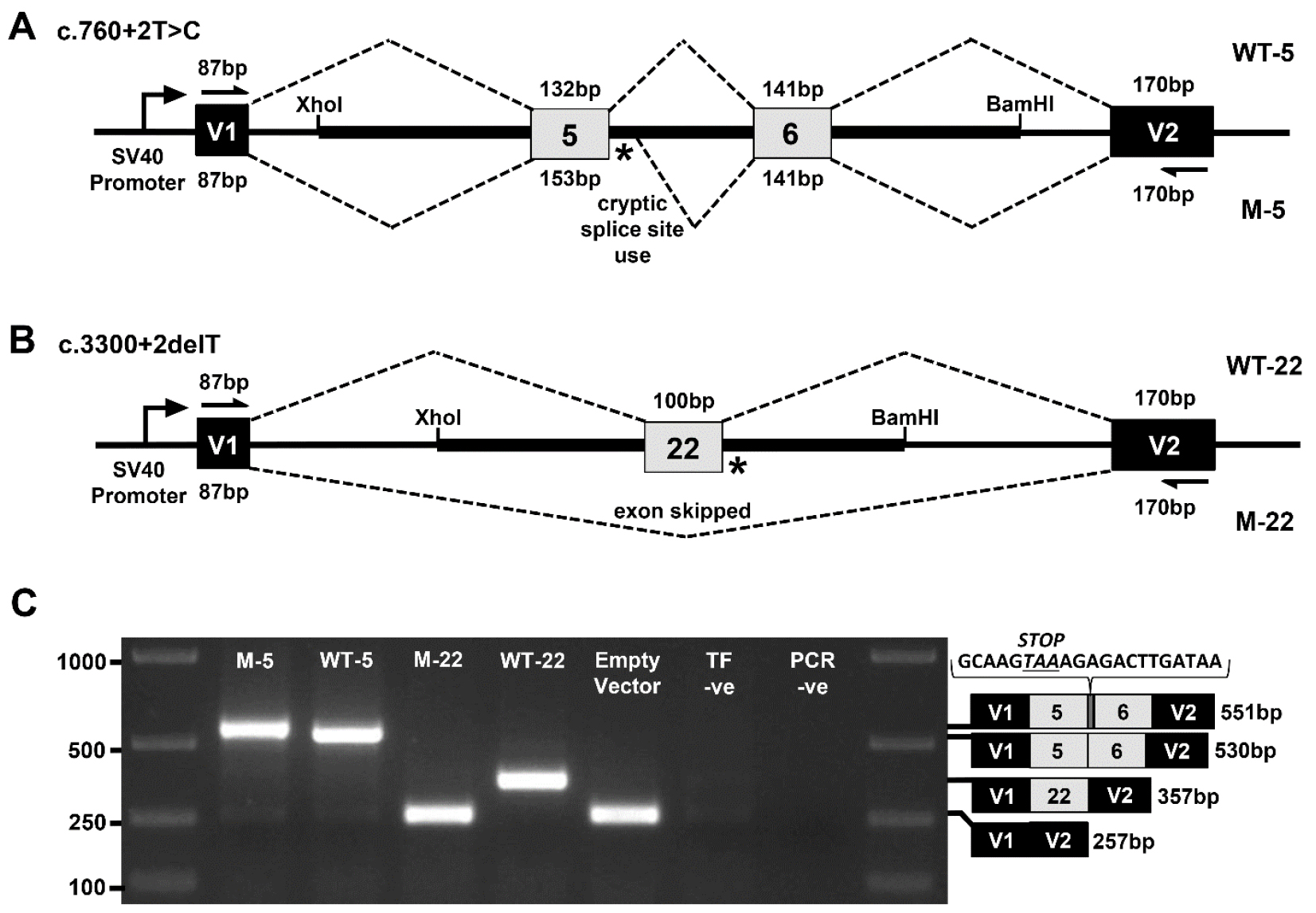
Figure 1. Exon trapping assay. Vector exons V1 and V2, are depicted as black boxes and TEK exons 5, 6, and 22 are shown in gray. Vector exon-specific primers are indicated by half-arrows in (A) and (B). Wild-type (WT) and mutant (M) splicing products, with included exon sizes in base pairs, are indicated by dashed lines above and below the construct, respectively. The locations of the splice site mutations are shown as an asterisk (*). A. Wild-type (WT-5) and mutant (M-5) genomic fragments containing TEK exons 5 and 6 were used to model the c.760+2T>C mutation. B. Wild-type (WT-22) and mutant (M-22) genomic fragments containing TEK exon 22 were used to model the c.3300+2delT mutation. C. Gel electrophoresis of RT-PCR products from transfected COS-7 cells. ‘Empty Vector’, cells transfected with vector containing no gDNA insert; ‘TF –ve’ (transfection negative), cells transfected with QIAGEN buffer EB only; ‘PCR –ve’ (PCR negative), PCR contamination control substituting water for cDNA template. Wild-type and mutant transcript content, determined by Sanger sequencing, is depicted to the right of the gel image. The additional 21 bp of intron 5 sequence identified within the M5 transcript is shown incorporating a premature termination codon between exons 5 and 6.
Materials and Reagents
- PCR
- 0.2 ml PCR tubes with caps (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM12225 )
- 1.5 ml conical base screw cap tube (USA Scientific, catalog number: 1415-8399 )
- Human genomic DNA samples with and without splice site variant (20 ng/μl)
- TE buffer, 10 mM Tris 1 mM EDTA pH 8.0 (Sigma-Aldrich, catalog number: 93283 )
- Custom-synthesized oligonucleotide primers incorporating restriction endonuclease sites to the 5’ end (Integrated DNA Technologies; https://www.idtdna.com/site/order/oligoentry):
- TEK_E5-F: 5’-ctgactgaCTCGAGCACAGCTCCAGCCTGTAACCAT-3’
- TEK_E5-R: 5’-tcagtcagGGATCCTCGGAACTACTTGGGAGCCTGT-3’
- TEK_E22-F: 5’-ctgactgaCTCGAGATTCCAAGGCAAATGCTGCTCT-3’
- TEK_E22-R: 5’-tcagtcagGGATCCTTGACTCCCAGATCGGTACAGC-3’
Note: Genome-specific sequences are underlined, restriction sites are shown in BOLD and an extra 8 bp added to the 5’ end are shown in lowercase. 5’-CTCGAG-3’ is the recognition sequence for XhoI and 5’-GGATCC-3’ is the recognition sequence for BamHI. ‘-F’ and ‘-R’ refers to the forwards and reverse primers in a pair, respectively. Resuspend primers and make 10 μM stocks with TE buffer.
- Phusion Hot Start II High-Fidelity DNA polymerase (Thermo Fisher Scientific, Thermo ScientificTM, catalog number: F549S )
- Molecular grade water, nuclease-free (Dot Scientific, catalog number: DS248700 )
- dNTP Set (QIAGEN, catalog number: 201913 )
- 2 mM dNTPs (see Recipes)
- 0.2 ml PCR tubes with caps (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM12225 )
- Gel electrophoresis
- 1 M Trizma hydrochloride solution, Tris-HCl, pH 7.8 (Sigma-Aldrich, catalog number: T2569 )
- 3 M sodium acetate buffer solution, pH 5.2 (Sigma-Aldrich, catalog number: S7899 )
- 0.5 M EDTA, pH 8.0 (Santa Cruz Biotechnology, catalog number: sc-203932 )
- Agarose (IBI Scientific, catalog number: IB70042 )
- SYBR safe DNA gel stain (Thermo Fisher Scientific, InvitrogenTM, catalog number: S33102 )
- Ficoll-400 (Dot Scientific, catalog number: DSF10400-25 )
- Bromophenol blue, sodium salt (MP Biomedicals, catalog number: 02152506 )
- Orange G (Sigma-Aldrich, catalog number: O3756 )
- HyperLadder 1 kb (Bioline, catalog number: BIO-33053 )
- 1x TAE buffer (see Recipes)
- 1% or 1.5% agarose gel (see Recipes)
- 10x gel loading dye (see Recipes)
- 1 M Trizma hydrochloride solution, Tris-HCl, pH 7.8 (Sigma-Aldrich, catalog number: T2569 )
- Cloning
- 1.5 ml Eppendorf tubes (VWR, catalog number: 20170-022 )
- Petri dishes, 100 x 15 mm (VWR, catalog number: 25384-302 )
- Whatman GD/X syringe filters, 0.2 μm pore size (Whatman, catalog number: 6901-2502 )
- BD 60 ml syringes, Luer-Lok Tip (BD, catalog number: 309653 )
- Pasteur glass pipettes, 230 mm (for spreading bacteria on plates) (WHEATON, catalog number: 357335 )
- 2 ml cryovial, self-standing (Simport, catalog number: T310-2A )
- 50 ml polypropylene conical tube, Falcon (Corning, Falcon®, catalog number: 352070 )
- 0.5 ml PCR tubes (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM12275 )
- JM109 E. coli competent cells (Promega, catalog number: L2001 )
- pSPL3 vector (Thermo Fisher Scientific, Invitrogen)
- Restriction endonuclease, XhoI (New England Biolabs, catalog number: R0146S )
- Restriction endonuclease, BamHI (New England Biolabs, catalog number: R0136S )
- QIAquick PCR Purification Kit (QIAGEN, catalog number: 28104 )
- T4 DNA ligase (New England Biolabs, catalog number: M0202S )
- SOC medium (Thermo Fisher Scientific, InvitrogenTM, catalog number: 15544034 )
- Luria Bertani broth (Lennox) powder microbial growth medium (Sigma-Aldrich, catalog number: L3022 )
- Select Agar (Thermo Fisher Scientific, InvitrogenTM, catalog number: 30391023 )
- Carbenicillin, disodium salt (Dot Scientific, catalog number: DSC46000-5 )
- Glycerol (Sigma-Aldrich, catalog number: G5516 )
- QIAprep Spin Miniprep Kit (QIAGEN, catalog number: 27104 )
- Carbenicillin antibiotic, 1,000x (see Recipes)
- LB (Luria-Bertani) medium with carbenicillin antibiotic (see Recipes)
- LB agar with carbenicillin antibiotic plates (see Recipes)
- 1.5 ml Eppendorf tubes (VWR, catalog number: 20170-022 )
- Mammalian cell culture
- Serological pipettes
5 ml (Corning, Costar®, catalog number: 4487 )
10 ml (Corning, Costar®, catalog number: 4488 )
25 ml (Corning, Costar®, catalog number: 4489 ) - Transfer pipette, polyethylene, general purpose blood bank, bulb draw 1.9 ml, sterile (Sigma-Aldrich, catalog number: Z350699 )
- Pasteur glass pipettes, 5.75” (for aspiration) (VWR, catalog number: 14673-010 )
- 25 cm2 (T25) cell culture flasks with 0.2 μm vent caps (Corning, catalog number: 430639 )
- COS-7 mammalian cells (ATCC, catalog number: CRL-1651 )
- Dulbecco’s modified Eagle’s medium, DMEM, + GlutaMAX-1 cell culture medium (Thermo Fisher Scientific, GibcoTM, catalog number: 10569010 )
- Fetal bovine serum, FBS (Thermo Fisher Scientific, GibcoTM, catalog number: 10437028 )
- Phosphate-buffered saline, PBS, pH 7.4, 1x (Thermo Fisher Scientific, GibcoTM, catalog number: 10010023 )
- Penicillin-streptomycin, 10,000 U/ml (Thermo Fisher Scientific, GibcoTM, catalog number: 15140122 )
- Trypsin-EDTA (0.5%), no phenol red (Thermo Fisher Scientific, GibcoTM, catalog number: 15400054 )
- FuGENE 6 Transfection Reagent (Promega, catalog number: E2691 )
- Opti-MEM I reduced serum medium (Thermo Fisher Scientific, GibcoTM, catalog number: 31985062 )
- Serological pipettes
- RNA extraction
- RNeasy Mini Kit (QIAGEN, catalog number: 74104 )
- QIAshredder (disposable cell-lysate homogenizers) (QIAGEN, catalog number: 79654 )
- RNeasy Mini Kit (QIAGEN, catalog number: 74104 )
- cDNA generation
- High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4374966 )
- High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Thermo Fisher Scientific, Applied BiosystemsTM, catalog number: 4374966 )
- RT-PCR
- 0.2 ml PCR tubes with caps (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM12225 )
- HotStarTaq DNA polymerase (QIAGEN, catalog number: 203203 )
- TE buffer, 10 mM Tris 1 mM EDTA pH 8.0 (Sigma-Aldrich, catalog number: 93283 )
- Vector-specific oligonucleotide primers (Integrated DNA Technologies; https://www.idtdna.com/site/order/oligoentry):
- V1-F: 5’-TCTGAGTCACCTGGACAACC-3’
- V2-R: 5’-ATCTCAGTGGTATTTGTGAGC-3’
Note: Resuspend primers and make 10 μM stocks with TE buffer.
- V1-F: 5’-TCTGAGTCACCTGGACAACC-3’
- 2 mM dNTPs (see Recipes)
- 0.2 ml PCR tubes with caps (Thermo Fisher Scientific, InvitrogenTM, catalog number: AM12225 )
- Sanger sequencing
- Sanger sequencing (GeneWiz commercial sequencing service; https://www.genewiz.com/en/Public/Services/Sanger-Sequencing)
- Sequencing primers (V1-F and V2-R oligonucleotide primers, same as for RT-PCR)
- Sanger sequencing (GeneWiz commercial sequencing service; https://www.genewiz.com/en/Public/Services/Sanger-Sequencing)
Equipment
- Gel doc system (Labnet International, model: EnduroTM GDS )
- Water bath (37 °C) (Fisher Scientific, model: Fisher ScientificTM IsotempTM 215 )
- Bacterial shaking incubator (37 °C) (GeneMate, model: Incubated Shaker Mini )
- Bacterial incubator (37 °C) (Lab-Line Imperial III Incubator)
- Mammalian cell incubator (37 °C, 5% CO2) (Eppendorf, model: Galaxy® 170 S )
- Bunsen Burner, simple natural gas burner version (Fisher Scientific, catalog number: S95941 )
- NanoDrop Lite spectrophotometer (Thermo Fisher Scientific)
- Eppendorf centrifuge (benchtop) (Eppendorf, model: 5415 D )
- PCR thermocycler (Eppendorf, model: Mastercycler® Pro S )
- Pyrex bottle/flask, 500 ml
- HotPlate Stirrer (GeneMate, model: HotPlate Magnetic Stirrer )
- VWR Spinbar Magnetic Stir Bar, Polygon with Pivot Ring, 6 mm (¼") diameter, 35 mm (1⅜") length (VWR, catalog number: 74950-290 )
- Fisher Vortex Genie 2 (Thermo Fisher Scientific, catalog number: 12-812 )
- Household microwave oven, 1,000 W
- Gel electrophoresis tank (and tray) (Bio-Rad Laboratories, model: Wide Mini-Sub GT Cell )
- Electrophoresis power pack (Bio-Rad Laboratories, model: PowerPacTM Basic Power Supply )
- Autoclave (Medium Healthcare Sterilizer) (Tuttnauer, model: 6690 )
- Biological safety cabinet (The Baker Company, model: SterilGARD e3 High Efficiency SG403A-HE )
- Handheld aspirator and collector trap (Argos Technologies, model: EV514 )
Software
- Sequencher software (version 5.2.4, Gene Codes Corporation; http://www.genecodes.com/sequencher)
- Primer3Plus (http://primer3plus.com/cgi-bin/dev/primer3plus.cgi)
Procedure
文章信息
版权信息
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Tompson, S. W. and Young, T. L. (2017). Assaying the Effects of Splice Site Variants by Exon Trapping in a Mammalian Cell Line. Bio-protocol 7(10): e2281. DOI: 10.21769/BioProtoc.2281.
分类
分子生物学 > DNA > DNA 克隆
分子生物学 > DNA > PCR
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。