

Virtual Screening of Transmembrane Serine Protease Inhibitors
跨膜丝氨酸蛋白酶抑制剂的虚拟筛选
(*contributed equally to this work) 发布: 2017年04月20日第7卷第8期 DOI: 10.21769/BioProtoc.2246 浏览次数: 10165
评审: HongLok LungShyam SrivatsTim Andrew Davies SmithVanesa Olivares-Illana
参见作者原研究论文
The authors used this protocol in:

Apr 2016
Advertisement










Abstract
The human family of type II transmembrane serine proteases includes 17 members. The defining features of these proteases are an N-terminal transmembrane domain and a C-terminal serine protease of the chymotrypsin (S1) fold, separated from each other by a variable stem region. Recently accumulated evidence suggests a critical role for these proteases in development of cancer and metastatic capacity. Both the cancer relevance and the accessibility of the extracellularly oriented catalytic domain for therapeutic and imaging agents have fueled drug discovery interest in the type II class of transmembrane serine proteases. Typically, the initial hit discovery processes aim to identify molecules with verifiable activity at the drug target and with sufficient drug-like characters. We present here protocols for structure-based virtual screening of candidate ligands for transmembrane serine protease hepsin. The methods describe use of the 3D structure of the catalytic site of hepsin for molecular docking with ZINC, which is a molecular database of > 30 million purchasable compounds. Small candidate subsets were experimentally tested with demonstrable hits, which provided meaningful cues of the ligand structures for further lead development.
Keywords: Molecular modeling (分子建模)Background
Controlled proteolytic activity plays a fundamental role in cellular processes and signaling, as evidenced by the presence of proteases in all organisms, including viruses, prokaryotes and eukaryotes. Not surprisingly, aberrantly regulated protease activity is causal to wide variety of human pathologies such as cardiovascular and inflammatory diseases, osteoporosis, neurological disorders and cancer (Turk, 2006; Bachovchin and Cravatt, 2012). In particular, development of primary cancer and metastatic capacity has been linked to several different classes of proteases including matrix metalloproteinases (MMPs), cysteine proteases (cathepsins) and membrane-associated serine proteases (Lopez-Otin and Matrisian, 2007). Recent clinical, genetic and functional data, suggesting a critical role for membrane-associated serine proteases in solid cancers, including cancer of prostate, ovarian and breast, have prompted new interest in the development of small molecule serine protease inhibitors for the treatment of cancer. Hepsin is a type II transmembrane serine protease and an attractive target for serine protease drug development due to frequent hepsin overexpression in common solid cancers, such as prostate and breast cancer, confinement of its overexpression on the membranes of cancer cells and due to positioning of the catalytic domain to the extracellular, i.e., more reachable, side of the cells (Antalis et al., 2010).
We describe here protocols for structure-based virtual screening of serine protease inhibitors using the catalytic site of hepsin for docking with drug-like subset of ZINC database. On the basis of virtual screening results, altogether 24 candidate compounds were purchased for further biochemical validation. Cell-free ELISA-based fluorogenic enzymatic assay using recombinant hepsin and fluorogenic peptide substrate Boc-Gln-Arg-Arg-AMC (BACHEM) was used for experimental validation and 30% inhibition of peptidolytic activity was set as threshold. Cut-off value was based on a notion that with the initial high micromolar concentration of compounds more than 30% inhibition would be required to determine reasonable IC50 value (Goswami et al., 2015; Tervonen et al., 2016) (Figures 1A and 1B). With these criteria, 3 out of 24 tested compounds showed inhibition potency (Tervonen et al., 2016). In conclusion, even though structure-based virtual screening is often considered as a complementary drug screening approach, the protocols reported here allowed us to demonstrate the feasibility and provided meaningful structural scaffolds for further development of specific serine protease inhibitors. However, it is important to stress that virtual screening hits typically do not demonstrate high potency, which is also true to the present screen. High-affinity ligands typically become available only after skillful medicinal chemistry optimization of selected hit structures.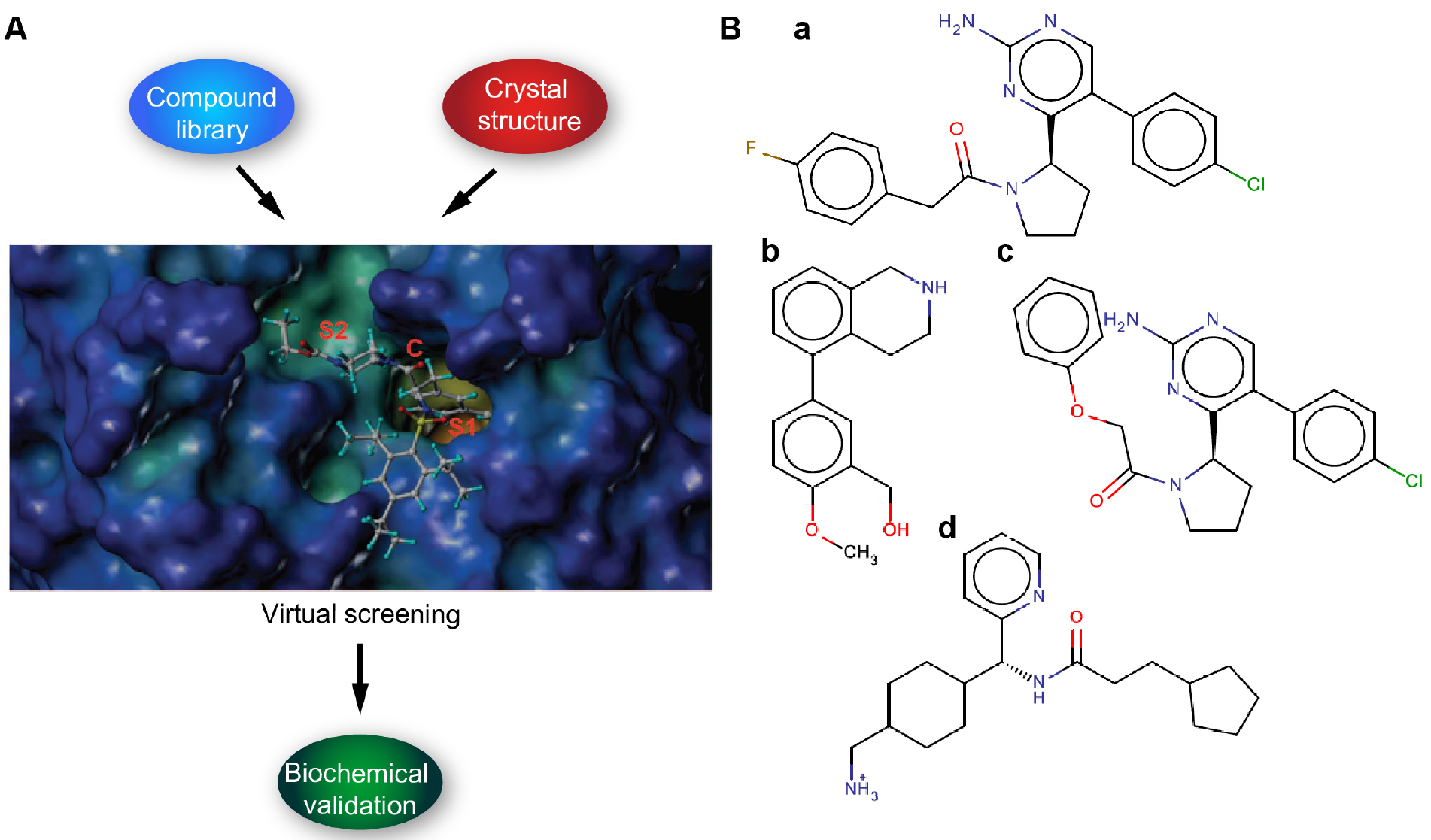
Figure 1. Schematic figure illustrating the workflow. A. Virtual screening (compound) library is prepared and the target protein structure analyzed and prepared. The virtual screening is carried out by docking (Glide SP/XP) and the putative binders are validated by in vitro biochemical assays. B. Examples of candidate ligands (a-d).
The virtual screening protocol presented here is a modified version from the original protocols described in our recent research paper (Tervonen et al., 2016). The modifications make the protocol fully compatible with the most updated versions of ZINC database and Schrödinger software. While the current virtual screening protocol is tailored for users of Schrödinger software and ZINC database, the protocol is versatile and also compatible with other molecular modeling packages. However, implementation of the present virtual screening protocol with other modeling packages would require a careful platform-specific validation of the docking settings. As a guidance, we provide a general overview of the validation procedures used in the present screen.
Materials and Reagents
- ViewPlate-96 black assay plates (PerkinElmer, catalog number: 6005225 )
- Recombinant human hepsin protein (R&D Systems, catalog number: 4776-SE-010 )
- DMSO (Sigma-Aldrich, catalog number: D8418 )
- Fluorogenic peptide substrate Boc-Gln-Arg-Arg-AMC (Bachem, catalog number: I-1655.0025 )
- Small-molecule compounds were purchased either from Asinex Corporation (Winston-Salem, NC, http://www.asinex.com) or MolPort (Riga, Latvia, https://www.molport.com/shop/index)
- WX-UK1 was a kind gift from Dr. Ramachandra (Aurigene Discovery Technologies Limited, Bangalore, India), however, it is also commercially available (for example, AURUM Pharmatech, catalog number: Z-3200 )
- Trizma base (Tris) (Sigma-Aldrich catalog number: T1503 )
- Calcium chloride (CaCl2) (Sigma-Aldrich, catalog number: 499609 )
- Brij-35 (Brij L23 30% solution in H2O) (Sigma-Aldrich, catalog number: B4184 )
- Sodium chloride (NaCl) (Sigma-Aldrich, catalog number: 31434-M )
- Activation buffer for recombinant human hepsin protein (see Recipes)
- Assay buffer for recombinant human hepsin protein (see Recipes)
Equipment
- ELISA plate reader FLUOstar Omega (BMG LABTECH, model: FLUOstar Omega )
- Computer hardware provided by CSC - IT Center for Science Ltd (Espoo, Finland)
Software
- Modeling software
Schrödinger Suite (Schrödinger, LLC, New York, NY) with Protein Preparation Wizard (Sastry et al., 2013), LigPrep, MacroModel and Glide (Friesner et al., 2004 and 2006; Halgren et al., 2004) modules, and a protocol version 2016-2 (https://www.schrodinger.com/suites/small-molecule-drug-discovery-suite) - Databases
- Screening and validation: ZINC database (downloadable via http://zinc15.docking.org/). ZINC database includes over 100 million purchasable compounds in 3D formats (Sterling and Irwin, 2015)
- Protein structure database (http://www.rcsb.org/pdb/home/home.do)
Procedure
文章信息
版权信息
© 2017 The Authors; exclusive licensee Bio-protocol LLC.
如何引用
Poso, A., Tervonen, T. and Klefström, J. (2017). Virtual Screening of Transmembrane Serine Protease Inhibitors. Bio-protocol 7(8): e2246. DOI: 10.21769/BioProtoc.2246.
分类
癌症生物学 > 癌症生物化学 > 蛋白质
生物化学 > 蛋白质 > 活性
您对这篇实验方法有问题吗?
在此处发布您的问题,我们将邀请本文作者来回答。同时,我们会将您的问题发布到Bio-protocol Exchange,以便寻求社区成员的帮助。